The Future is Here: BioMarin’s Roctavian First Hemophilia A Gene Therapy Paving the Way for a Cure
Jul 14, 2023
Table of Contents
Hemophilia A is a rare genetic bleeding disorder that primarily affects males. Hemophilia A is the most common type of hemophilia, accounting for approximately 80% of all cases. It is estimated to occur in about 1 in 5,000 to 10,000 male births. As per the Centers for Disease Control (CDC) (2020), the estimated prevalence of hemophilia in the United States is 12 cases per 100,000 US males for hemophilia A.
According to DelveInsight’s analysis, the total number of prevalent cases of hemophilia A was found to be ~45K in the 7MM in 2021. Among the 7MM, the US accounted for the highest patient share of hemophilia A, followed by EU4 and the UK, and Japan. As per the estimates, severe cases of hemophilia A are more prominent in comparison to mild and moderate cases.
Downloads
Article in PDF
Recent Articles
- Plasma Proteins Therapeutics: Transforming Treatment in Hematology and Beyond
- Opportunities and Challenges for Cell and Gene Therapies
- Hemophilia A- Market Scenario
- AAV Vectors in Gene Therapy: How Recent Clinical Advances are Unraveling New Potentials?
- Novo Nordisk’s Concizumab for Hemophilia; AbbVie Ends its Alliance with Alector; ADC Therapeutics...
Currently, there is no cure, but researchers are actively engaged in finding the cure through gene therapy. One hope is that by inserting a healthy version of the defective blood factor gene, a person with hemophilia will be able to produce reasonable amounts of a factor on their own.
HEMLIBRA: An Effective Hemophilia A Treatment
HEMLIBRA (Emicizumab-kxwh injection), a product of “Genentech/Chugai/Roche”, is a novel bispecific antibody that was first approved in 2018 for patients with hemophilia A with FVIII inhibitors in the US by the US Food and Drug Administration (FDA). However, in 2018, HEMLIBRA was approved for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children, ages newborn and older, with hemophilia A without factor VIII inhibitors (FVIII).
In addition to this, in December 2018, Chugai obtained regulatory approval for HEMLIBRA from the Ministry of Health, Labour, and Welfare for prophylactic treatment for people with hemophilia A without inhibitors to factor VIII, as well as for additional dosage and administration as biweekly or every four-week treatment for people with hemophilia A with inhibitors to factor VIII. Later on, in March 2019, the European Commission (EC) also approved HEMLIBRA for people with severe hemophilia A without factor VIII inhibitors. The EU Commission also approved that HEMLIBRA can be used with multiple dosing options(once weekly, every two weeks, or every four weeks) for all indicated people with hemophilia A, including those with factor VIII inhibitors.

HEMLIBRA has been studied in one of the largest pivotal clinical trial programs in people with hemophilia with and without factor VIII inhibitors, including four Phases III studies (HAVEN 1, HAVEN 2, HAVEN 3 andHAVEN 4). This approval was based on results from the pivotal HAVEN 3 and HAVEN 4 studies.
FDA Approval of First Gene Therapy for Hemophilia A Treatment: BioMarin’s Roctavian
After an initial rejection in 2020 and a review delay earlier this year, BioMarin’s Roctavian has finally received FDA approval to launch a gene therapy for a not-so-rare disorder. On June 29, 2023, BioMarin announced that the FDA had approved Roctavian as a one-time treatment for patients with severe hemophilia A. The announcement comes seven months after the FDA approved CSL Behring’s hemophilia B gene therapy Hemgenix, which has a list price of $3.5 million and is now the most expensive drug in the world.
Roctavian, a one-time single-dose infusion, is exclusively allowed for use in individuals who do not have antibodies against adeno-associated virus serotype 5 (AAV5), as established by an FDA-approved test. Roctavian has a wholesale acquisition cost, or WAC, of $2.9 million for the average patient for this one-time, single-dose treatment. ROCTAVIAN was approved by the European Medicines Agency for the first time in August 2022.
“Adults with severe hemophilia A face a lifelong burden, with frequent infusions and a high risk of health complications, including uncontrolled bleeding and irreversible joint damage,” said Dr. Steven Pipe, an investigator in the Phase III study and a professor of pediatrics and pathology at the University of Michigan. “ROCTAVIAN’s approval as the first gene therapy for severe haemophilia A has the potential to transform the way we treat adults based on years of bleed control after a single, one-time infusion.”
The FDA approval is based on data from the global Phase III GENEr8-1 study, the biggest Phase III trial of any gene therapy in hemophilia. Of the 134 patients who got ROCTAVIAN in the research, 112 had baseline annualized bleeding rate (ABR) data gathered prospectively throughout at least six months of FVIII prophylaxis prior to starting ROCTAVIAN. The remaining 22 patients had their baseline ABR recorded retrospectively. All patients were followed for at least three years.
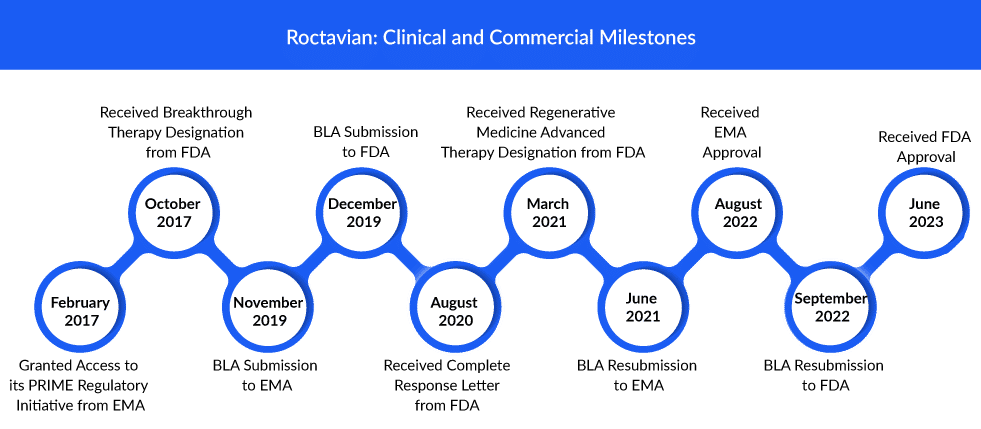
According to the FDA-approved labeling for ROCTAVIAN, the 112 patients who had their 6-month baseline ABR collected prospectively experienced a 52% reduction in ABR after receiving ROCTAVIAN (2.6 bleeds/year) through the end of follow-up (median of three years) compared to their baseline ABR while receiving routine FVIII prophylaxis (5.4 bleeds/year). This conclusion was based on an FDA investigation that imputed an ABR of 35 in 13 individuals throughout their prophylactic periods. These patients also reported a significant decrease in the rate of spontaneous and joint bleeds following ROCTAVIAN treatment (observed mean ABR of 0.5 bleeds/year for spontaneous bleeds and 0.6 bleeds/year for joint bleeds) compared to their baseline rate while receiving routine FVIII prophylaxis (observed mean ABR of 2.3 bleeds/year for spontaneous bleeds and 3.1 bleeds/year for joint bleeds).
Without the use of frequent prophylaxis, the majority of research subjects continued to respond to treatment through year three and beyond. BioMarin will continue to evaluate the treatment’s long-term effects with an extension study that will follow all clinical trial participants for up to 15 years, as well as post-approval studies that will follow individuals dosed in a real-world scenario for 15 years or more.
Other Hemophilia A Gene Therapy Competitors in Pipeline
Key players such as Pfizer, Roche, Spark Therapeutics, Sangamo Therapeutics, ASC Therapeutics, and others, are involved in developing gene therapies for hemophilia A treatment. The expected launch of these therapies will lead to a significant increase in the hemophilia A treatment market size during the forecast period (2023–2032).
Sangamo in collaboration with Pfizer is investigating Giroctocogene fitelparvovec, which is a recombinant adeno-associated virus serotype 6 vector (AAV6) encoding the complementary deoxyribonucleic acid for B domain deleted human FVIII. Currently, the company is conducting Phase III trials to attain better and clear results about the efficacy of this candidate. The FDA lifted its clinical hold on the Phase III study Affine for the hemophilia A gene therapy giroctocogene fitelparvovec in March. Patient dosing resumed in November 2022 in the AFFINE trial. In September 2022, Pfizer and Sangamo Therapeutics announced that the Phase III AFFINE study evaluating giroctocogene fitelparvovec has re-opened recruitment. Hemophilia A pivotal data readout is expected by the first half of 2024, and a BLA submission is anticipated by the second half of 2024.
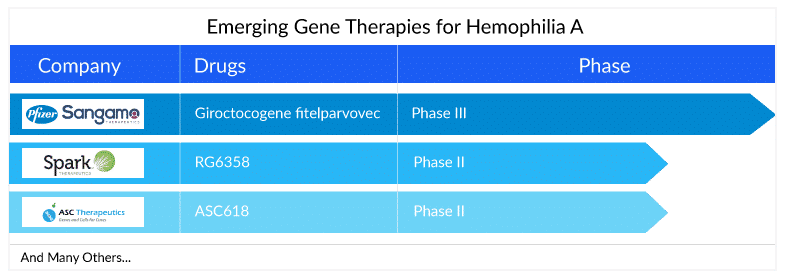
Apart from this, RG6357 (SPK-8011), which is under development by Roche’s subsidiary Spark Therapeutics is an intravenously administered novel bio-engineered AAV vector utilizing the AAVLK03 capsid, also referred to as Spark200. Currently, a Phase I/II clinical research study is being conducted by the company to determine the safety and efficacy of the factor VIII gene transfer treatment with SPK-8011 in individuals with hemophilia A.
In addition to this, Spark Therapeutics is also developing RG6358 (also known as SPK-8016) a novel, internally developed investigational gene therapy for hemophilia A, or Factor VIII deficiency, to address the inhibitor market using a gene-based approach. It aims to boost factor VIII levels in the blood of hemophilia A patients who develop inhibitors. In ASH 2022, the company announced data from its Phase I/II clinical trial of investigational SPK-8011 in hemophilia A. In the updated safety analysis, no new safety signals were reported related to investigational SPK-8011; no deaths, thrombotic events, or FVIII inhibitor development were reported.
ASC Therapeutics is also evaluating ASC618, a second-generation gene therapy for hemophilia A treatment. ASC618 has a unique liver-specific promoter and a bioengineered, codon-optimized B domain-deleted FVIII variant; in preclinical experiments, ASC618 displays a 10-fold increase in FVIII production and secretion when compared to native human FVIII bioengineered gene constructs. ASC618 can potentially improve the durability of clotting factor biosynthesis and release by reducing cellular stress and inducing the unfolded protein response, which may result in decreased FVIII production by liver cells.
ASC Therapeutics currently conducting phase I/II clinical research to assess ASC618’s safety, tolerability, and preliminary efficacy. The US Food and Drug Administration granted the program IND clearance, as well as Fast-Track and Orphan Drug Designations. The Committee for Orphan Medicinal Products (COMP) of the European Medicines Agency (EMA) has also granted an Orphan Medical Product Designation.
Pioneering Paths to a Brighter Future in the Hemophilia A Treatment Space
The future outlook of hemophilia A treatment is exceptionally promising, with groundbreaking advancements set to transform the lives of individuals affected by this condition. Gene therapy, extended half-life therapies, non-replacement treatments, personalized hemophilia A medicine, and improved management strategies offer a multifaceted approach to addressing the challenges associated with hemophilia A. As research and innovation continue to unfold, there is a renewed sense of hope that the hemophilia community will witness significant improvements in treatment efficacy, safety, and overall quality of life in the years to come.
FAQs
Hemophilia A is a genetic bleeding disorder in which a person lacks or has low levels of clotting factor VIII proteins. Blood cannot clot properly to control bleeding if factor VIII levels are low. Even though it is passed down from parents to children, approximately one-third of cases discovered have no prior family history.
The severity and hemophilia A symptoms can vary greatly between individuals. Hemophilia A can be mild, moderate, or severe. In these categories, factor VIII levels are 5-40% of normal, 1-5%, and less than 1%, respectively.
Genetic inheritance is the cause of hemophilia A. It is caused by an X-linked recessive trait, with the defective gene on the X chromosome. Females have two X chromosome copies. So, if the factor VIII gene on one chromosome fails, the gene on the other chromosome can produce enough factor VIII.
Screening tests and clotting factor tests are used in the hemophilia A diagnosis. Screening tests are blood tests that determine whether or not the blood is clotting properly. Clotting factor tests, also known as factor assays, are required to determine the cause of a bleeding disorder. This blood test determines the type and severity of hemophilia.

Downloads
Article in PDF
Recent Articles
- FDA Approves PENBRAYA for Most Common Serogroups Causing Meningococcal Disease; BIMZELX Approved ...
- Opportunities and Challenges for Cell and Gene Therapies
- First Gene Therapy for Severe Hemophilia A; FDA Approves CellTrans’s Type 1 Diabetes Cellular The...
- AAV Vectors in Gene Therapy: How Recent Clinical Advances are Unraveling New Potentials?
- Global Hemophilia Market



