Opportunities and Challenges for Cell and Gene Therapies
Feb 05, 2024
Table of Contents
Gene therapy is an innovative medical approach that manipulates an individual’s genetic material to prevent or treat diseases. It primarily involves introducing, modifying, or substituting genes within a patient’s cells. The central objective of gene therapy is to rectify genetic abnormalities, insert missing genes, or regulate gene activity, thereby addressing the underlying causes of various health conditions. This is accomplished by delivering specific genetic material, often DNA, into the patient’s cells using diverse methods like viral vectors or gene-editing tools like CRISPR-Cas9. Gene therapy exhibits the potential to address an extensive spectrum of medical ailments, encompassing genetic disorders such as cystic fibrosis and muscular dystrophy, as well as acquired diseases and specific forms of cancer. A major advantage of gene therapy is that instead of continuous administration of a drug with a relatively short half-life, gene therapy allows the continuous expression of genes after a single treatment.
Cell therapy transfers autologous or allogeneic cellular material into a patient for medical purposes. Cell therapy constitutes another pioneering medical approach that entails introducing viable and functional cells into a patient to restore or augment the function of damaged or diseased tissues. This strategy is advantageous when cell dysfunction or loss significantly contributes to developing a particular ailment. The cells employed in cell therapy may be obtained from the patient’s own body (autologous) or a compatible donor (allogeneic). Several cell therapies exist, including stem cell therapy, CAR-T cell therapy, and tissue engineering. For instance, stem cell therapy involves utilizing stem cells to replace or mend impaired tissues, rendering it applicable for treating conditions like blood disorders and organ damage.
Downloads
Article in PDF
Recent Articles
- CAR T-Cell Therapies in Non-Hodgkin’s Lymphoma Treatment: A Revolutionary Approach
- AAV Vectors in Gene Therapy: How Recent Clinical Advances are Unraveling New Potentials?
- Bristol-Myers Squibb to nix; Pfizer commits $100M; Merck and GSK production issues; Shire unloads...
- Gene Therapies as a Game-Changer in Ophthalmology: Eyeing the Future
- Notizia
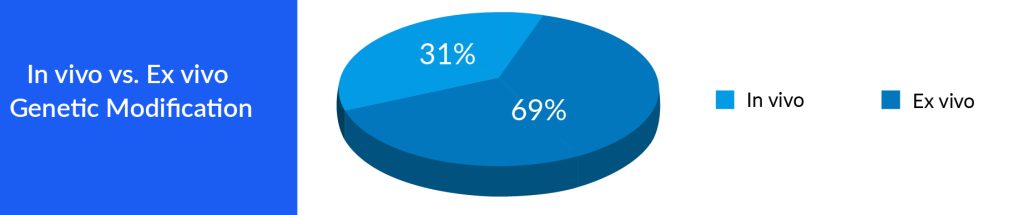
Both gene therapy and cell therapy represent notable advancements in the realm of medical science, offering substantial potential for the treatment of an extensive array of diseases. These therapeutic approaches are actively investigated and developed for conditions such as genetic disorders, cancer, autoimmune ailments, and neurodegenerative disorders. As researchers continue to refine these methodologies and address their associated challenges, gene therapy, and cell therapy hold the promise of profoundly influencing the future of medicine, presenting innovative treatments and even potential remedies for various incapacitating conditions. Nonetheless, the ongoing exploration and thorough consideration of ethical and safety considerations are crucial as these pioneering therapies advance toward broader clinical use. A major advantage of gene therapy is that continuing gene expression (s) allows for a cure following a single treatment instead of continuing to administer a drug with a relatively short half-life. One gene therapy strategy involves correcting an error in a gene or replacing a defective gene.
Opportunities for Cell and Gene Therapies
Potential for Success
Different clinical trials have demonstrated the efficacy of cell and gene therapy. The impact of these treatments will greatly increase even though the original approvals were for relatively limited patient groups because of the substantial pipeline of gene therapy studies for conditions including hemophilia and different types of blindness. The increasing number of trials is encouraging, particularly in light of the potential impact this technology may have on patients’ lives.
In humans, the inherited condition that best corresponds with the dogs’ vision loss is Leber Congenital Amaurosis (LCA). LCA hinders the proper response of the retina, a layer of light-sensitive cells located at the posterior part of the eye, to the impact of photons and the transmission of signals to the brain. This condition may lead to involuntary eye movements (nystagmus), the incapacity of pupils to react to light, and typically culminates in complete blindness by the age of 40. Scientists have associated the ailment with mutations or deletions in any 27 genes related to retinal development and function.
Mutations in RPE65 represent just one factor contributing to inherited retinal dystrophy, yet it was a factor that Bennett and Maguire were able to address. Utilizing a harmless adeno-associated virus (AAV), they programmed the virus to locate retinal cells and introduce a healthy gene variant. This modified virus was then injected directly beneath the retina of a patient’s eye. Following a series of clinical trials, the Food and Drug Administration approved in 2017 voretigene neparvovec-rzyl (marketed as Luxturna) as a treatment for any hereditary retinal dystrophy resulting from the mutated RPE65 gene, including LCA type 2 and retinitis pigmentosa, another congenital eye disorder affecting photoreceptors in the retina.
Due to the early success of these cell therapy initiatives, larger populations are now the focus of broader programs, which began with leukemia and are currently targeting lymphomas. Ultimately, multiple myeloma is the most difficult opportunity with the highest possibility for positive results. The notable advantage is that cell and gene therapy uses the body’s systems, either the cellular immune system or the ability to repair and replace defective or missing genes; hence, cell and gene therapies are considered better options for personalized medicine. CAR-T cell therapy is arguably among the most personalized medicines one can consider. The patient’s T cells are extracted, modified, activated, expanded, purified, and returned to the patient.
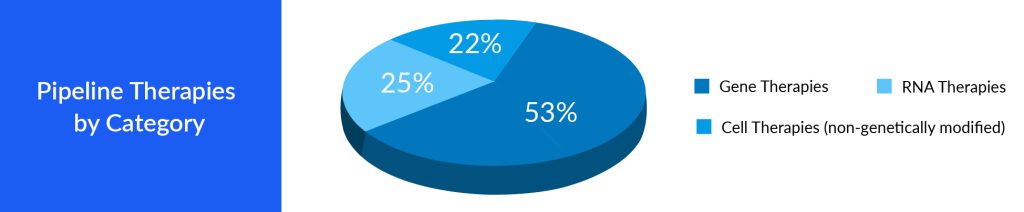
Faster Approval for Gene and Cell Therapies
Speedier approvals allow pharmaceutical companies to start generating revenue from these therapies sooner, essential for recouping the significant investments made in research and development. Companies that secure faster approvals gain a competitive advantage. They can establish a dominant position in the market, which can be challenging for later entrants to overcome. The recently approved ROCTAVIAN, intended for treating adults with severe hemophilia A, received RMAT designation in 2021 by the FDA. RMAT designation allows for accelerated approval based on surrogate or intermediate endpoints. RMAT goes beyond breakthrough therapy features by allowing for accelerated approval of drugs based on surrogate endpoints. Designations like these speed up the approval process of cell and gene therapy. Accelerated approvals provide pharmaceutical companies with a distinct competitive edge in the market. When a company secures accelerated approval for its gene or cell therapy, it gains a significant head start over its competitors, enabling it to establish a leading position. This advantage is particularly important in markets where numerous companies compete to introduce similar therapies. Companies that are the first to launch their therapies can secure a larger portion of the patient and healthcare provider base, often establishing the standard for the therapy’s adoption. The conventional approval pathways necessitate an approximate 18 months for the approval process, whereas the expedited pathway expedites the approval of cell and gene therapies within approximately 6 months.
Furthermore, this competitive advantage has the potential to evolve into long-lasting market leadership. Once a company is acknowledged as a reputable gene or cell therapy provider, it can foster brand recognition and customer allegiance. This can act as a deterrent to potential new entrants and create hurdles for subsequent competitors aiming to displace the initial market leaders. This competitive edge can lead to sustained revenue growth and profitability, further solidifying the company’s position within the 7MM.
Start-up Lead Innovation in Cell and Gene Therapy Space
Most gene and cell therapy innovators are small biotech start-ups or research universities. Sometimes, these companies collaborate with larger pharma companies with specialized targeted therapy. Small biotech start-ups typically exhibit greater flexibility and creativity than their larger pharmaceutical counterparts. They can quickly adjust to emerging technologies, groundbreaking scientific breakthroughs, and evolving market trends. Considering the fund’s availability in the current market scenario, these companies have the opportunity to raise funds and diversify their clinical programs across multiple therapeutic areas.
In 2017, Gilead announced the acquisition of Kite Pharma with USD 11.9 billion. At that time, Gilead realized the value of Yescarta and Kite’s pipeline, which includes engineered cell treatments for leukemia, multiple myeloma, cervical cancer, and solid tumors. In the realm of return on investment (ROI), the transaction proved financially advantageous for Gilead. In 2022, the annual sales of Gilead were reported at USD 26.9 billion, and the annual sales in the cell therapy space, including Yescarta and Tecartus, were reported at USD 1.5 billion. The revenue from Yescarta alone was reported at USD 1.1 billion.
In November 2023, AstraZeneca announced a collaboration and investment agreement with Cellectis, a clinical-stage biotechnology company, to accelerate the development of next-generation therapeutics in areas of high unmet need, including oncology, immunology, and rare diseases. Under the terms of the collaboration agreement, AstraZeneca will leverage the Cellectis proprietary gene editing technologies and manufacturing capabilities, to design novel cell and gene therapy products, strengthening AstraZeneca’s growing offering in this space. As part of the agreement, 25 genetic targets are exclusively reserved for AstraZeneca, from which up to 10 candidate products could be explored for development.
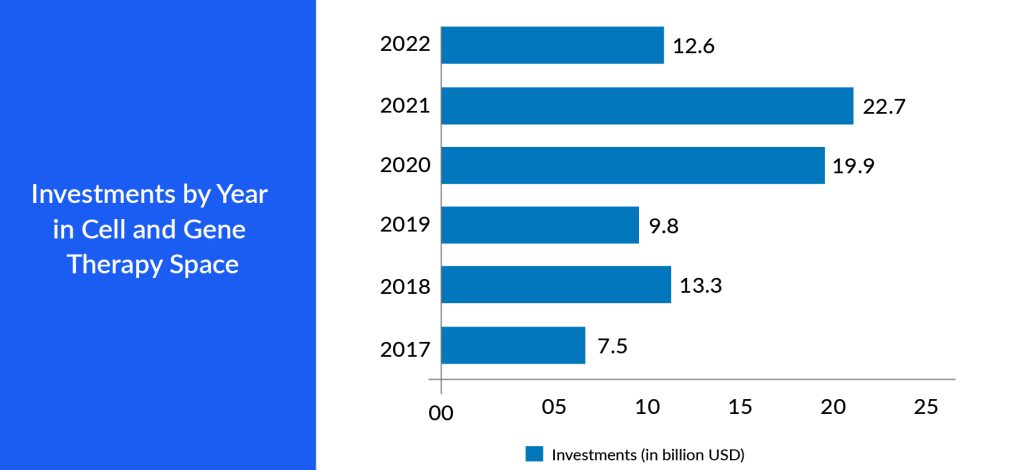
Big pharma companies strategically collaborate or acquire start-up pharma companies outright. This can provide them immediate access to a mature product, a promising pipeline, or a talented pool of researchers and scientists. Furthermore, these start-ups enrich the competitive landscape, encouraging advancements and cost-effectiveness in developing cell and gene therapies. As start-ups pioneer new approaches and forge collaborations with established pharmaceutical companies, navigating regulatory pathways within the seven major markets becomes easier. This competitive environment fosters an environment where the most promising therapies can be brought to market, benefitting patients and driving economic growth in these key pharmaceutical markets. This adaptability can lead to the creation of state-of-the-art treatments, capturing the interest of investors and patients in the 7MM.
Challenges for Cell and Gene Therapies
Manufacturing Challenge
The production of cell and gene therapies is highly complex, and manufacturing capacity is a major limiting factor in treating more patients. Cell and gene therapies generally involve a long process that can take weeks to complete as a patient’s cells are collected, multiplied, and modified in a laboratory. The manufacturing of viral vectors is very expensive because of low yields (approximately 10 doses per batch) due to low transfection efficiency.
A significant manufacturing difficulty is proving the product’s potency, safety, and quality because it requires high cost and skills to assemble the different components in a way that allows them to function. Discrepancies between batches in the production of cell and gene therapies arise primarily from the inherent biological intricacies and sensitivity of the involved living cells. These therapies commonly employ primary, stem, or genetically modified cells, which naturally display biological variability. Factors like cell growth rates, gene expression, and therapeutic efficacy may differ between batches due to the inherent diversity of cellular populations. The transition from laboratory-scale processes to large-scale manufacturing poses challenges in maintaining consistent culture conditions, nutrient distribution, and oxygen levels, contributing to variations in cell behavior across batches. Additionally, the use of viral vectors for gene delivery introduces complexity, as the stability and performance of these vectors can fluctuate, affecting the overall effectiveness and uniformity of the therapeutic product.
Challenges have also arisen from chemistry, manufacturing, controls, and quality. Foreign DNA following purification has resulted in multiple clinical trial holds. One of the main problems in this area is that the manufacturing process often involves manual labor, which is both time-consuming and expensive. These challenges affect the manufacturing and scale-up processes of cell and gene therapy in the 7MM.
High Cost Associated With Treatment
The high cost of authorized gene therapies is a significant hurdle for high-income countries (HICs), hindering their widespread availability in low to middle-income countries (LMICs). Gene therapies often come with steep price tags, reflecting the substantial investments in research and development, the complexity of their manufacturing processes, and their focus on addressing relatively small patient populations, particularly for rare diseases. In HICs, healthcare systems are generally better equipped to manage these elevated expenses due to their greater economic resources and comprehensive insurance coverage. Nevertheless, even in wealthier settings, the affordability of gene therapies can strain healthcare budgets and raise concerns about their long-term sustainability. For example, GLYBERA, the first gene therapy approved in Europe, was priced at EUR 1 million and was later withdrawn because no country provided coverage because of the cost. Hemgenix is recently approved to treat hemophilia B, a rare, lifelong bleeding disorder. The single-dose therapy is priced at USD 3.5 million, according to CSL Behring, which made it the most expensive drug at the time of FDA approval in November 2022.
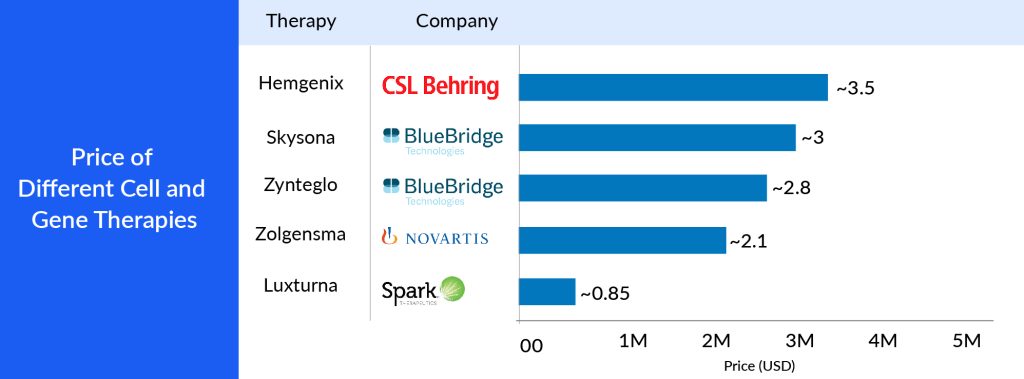
The challenge gets amplified when considering LMICs, where healthcare resources are more limited and a significant portion of the population lacks access to basic healthcare services, let alone expensive advanced treatments. The growing cost gap between HICs and LMICs exacerbates global healthcare disparities, making it increasingly difficult for LMICs to offer licensed gene therapies to their populations. These therapies hold significant potential for addressing severe and often disabling genetic disorders, but their current pricing structure makes it challenging to ensure equitable access in the 7MM.
The Logistical Challenge
As autologous cell therapies are highly sensitive, the therapy must be administered to the patient immediately after the cells are extracted. The manufacturing and delivery timings are crucial for patients in the late stages of a disease. Many raw materials required to produce the therapy are unconventional such as cells from patients, but the facilities that manufacture therapies are usually located away from the patient’s clinical setting and hence cause delays in deliverables.
Even the smallest logistical error can have serious consequences. The logistical precision required to produce autologous cell therapies is not feasible to achieve consistently and at scale using traditional manufacturing approaches. Chain of identity and chain of custody processes are also complex to create and manage. It is also very difficult to make even small changes in the manufacturing process after approval. SUPAC is scale-up and the post-approval changes (changes made after approval), like in the drug formulation, batch size, process, equipment, and manufacturing site.
Future Prospects and Conclusion
The future outlook for cell and gene therapy is promising and poised to bring a transformative shift in the medical landscape. These therapeutic approaches are anticipated to revolutionize disease treatment by targeting genetic and cellular origins. Advancements in precision medicine, particularly in genomics, are expected to drive the development of more tailored and personalized cell and gene therapies. This personalized strategy, exemplified in the success of CAR-T cell therapies for cancer, can provide symptomatic relief and curative interventions by addressing the root causes of diseases. Ongoing innovations in gene editing technologies such as CRISPR-Cas9 and improvements in manufacturing processes are likely to enhance the scalability and affordability of these therapies, making them accessible to a wider range of patients.
Furthermore, the intersection of regenerative medicine and cell therapy opens up new tissue repair and regeneration possibilities. Utilizing the body’s cells to mend damaged tissues and organs has profound implications, spanning conditions from cardiovascular diseases to neurodegenerative disorders. As regulatory frameworks adapt to the unique challenges posed by these novel therapies, collaborative efforts between academia, industry, and regulatory entities are expected to expedite the translation of cutting-edge research into approved and widely available treatments. With ongoing investments, technological breakthroughs, and an increasingly comprehensive understanding of the human genome, the future of cell and gene therapy holds the promise of ushering in a new era in medicine, where diseases are not merely managed but effectively cured at their molecular origins.
In conclusion, gene and cell therapy presents unparalleled opportunities, poised to revolutionize healthcare despite challenges such as complexities in manufacturing and scaling up, logistical issues, and the high associated costs. The increasing attention and investment in this field highlight its potential to revolutionize healthcare. As scientific progress continues to address these obstacles, the industry is making strides in overcoming them, heralding a future where personalized and targeted treatments become more accessible. Collaborative efforts among researchers, industry players, and regulatory bodies play a pivotal role in navigating the evolving dynamics of cell and gene therapy, ensuring the realization of their considerable promise and reshaping the healthcare paradigm with improved patient outcomes.

Downloads
Article in PDF
Recent Articles
- The Growing Burden Of Neurodegenerative Disorders
- Will The Burgeoning Gene Therapies Make a Difference in Dystrophic Epidermolysis Bullosa Patients...
- Notizia
- Break the Gene Therapy Barrier with DelveInsight’s Gene Therapy Reports
- Unveiling the Potential of TROP-2 Inhibitors: A New Frontier in Cancer Treatment



