Avrobio’s Gene Therapy for Cystinosis Treatment: A New Hope to Overcome the Existing Medical Unmet Needs
Nov 26, 2021
Table of Contents
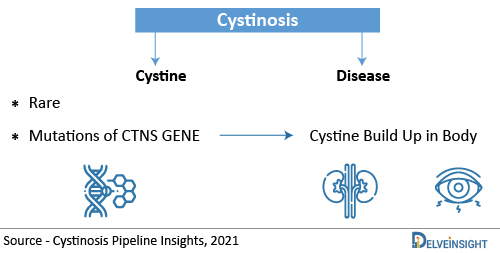
Cystinosis is a genetic condition that causes the buildup of the amino acid “cystine” in the cells of the body is known as cystinosis. Due to the excessive accumulation of cystine in various cells, organs, tissues, including the kidneys, eyes, liver, thyroid, muscles, pancreas, brain, and white blood cells of the body are damaged. Cystinosis can be classified as a lysosomal storage disorder, a rare, multisystem genetic disorder. Generally, Cystinosis is broken down into three different forms known as nephropathic cystinosis, intermediate cystinosis and non-nephropathic (or ocular) cystinosis. The kidneys and eyes are the two organs most often affected. Nephropathic Cystinosis presents in infancy and is the most common and severe form. Early detection and prompt treatment are critical in slowing the development and progression of symptoms associated with cystinosis.
Cystinosis – Causes, Types, Diagnosis, and Symptoms
Cystinosis is caused by mutations of the CTNS gene and is inherited as an autosomal recessive disease. The CTNS gene contains instructions for encoding a protein called cystinosin that is required to transport the amino acid cystine out of lysosomes and into the rest of a cell. Lysosomes degrade certain proteins into their component amino acids such as cystine. Cystine is then transported out of the lysosome by cystinosin. Deficient levels of functional cystinosin result in the accumulation of cystine in the lysosomes of various tissues and organs of the body. The accumulated cystine forms crystals, which eventually damage the affected organs. Cystinosis is not something that a person can catch from another individual. It is a genetic condition, which means a child is born with it. A child can get cystinosis if both parents are carriers of the disease. A child suffers from cystinosis when the specific gene that does not work right is passed down from both parents. A parent of a child with cystinosis carries one copy of the abnormal CTNS gene. The parents are carriers and have no signs of the disease. The genetic mutation causes a defect in the transport of cystine out of the cells. The cystine crystallizes in the cell and destroys cells.
Downloads
Article in PDF
Recent Articles
- DelveInsight’s Central Nervous System Disorders based Gene Therapy Reports
- VBL Therapeutics’ VB-111 (ofranergene obadenovec); BMS’s mavacamten (Camzyos); Merck’s KEYTRUDA; ...
- Snippet : Genetic Mutation
- Glioblastoma Multiforme: Advancements in the Treatment Paradigm of the Malignant Condition
- EMA to relocate to Amsterdam; Roche’s prospects; Amgen’s Humira Biosimilar delayed; Purdue’s opio...
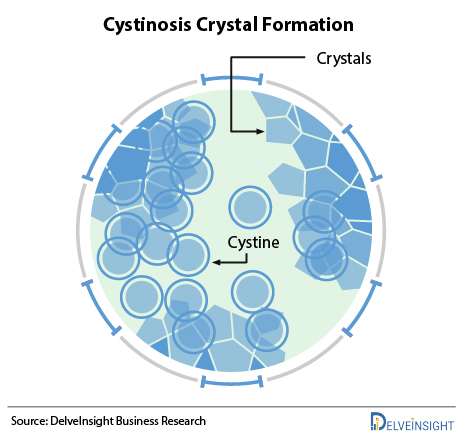
There are three types of Cystinosis, depending on when symptoms first appear (known as onset): Infantile (early-onset) Cystinosis; later childhood or Adolescent (late-onset) Cystinosis; and Adult cystinosis. About 95% of patients with cystinosis have the infantile/early-onset form, making it the most common variant of this disease, and most patients develop kidney problems. In many cases, the categorization is also done as, Nephropathic Cystinosis, Intermediate Cystinosis, and Non-nephropathic (or ocular) Cystinosis.
The diagnosis of Cystinosis is based upon the identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests. Due to the availability of specific cysteamine therapy, early diagnosis and management of cystinosis have a great impact on the clinical outcome of patients. There are three main diagnostic modalities for Cystinosis. The current gold standard is the detection of elevated cystine levels in white blood cells (WBCs), being extremely sensitive and precise for Cystinosis disease. Molecular testing of the relatively small CTNS gene (12 exons but only 10 are codings) is also a well-established technique revealing 95% of disease-causing mutations. The third clinically used confirmatory option is the detection of the characteristic corneal cystine crystals by slit-lamp examination. Being a monogenic disease, molecular diagnosis is efficient as a confirmatory tool; however, it is usually more time-consuming than cystine measurement. In about five percent of patients, pathogenic mutations are not easily discovered by the usual CTNS gene sequencing, being either deeply intronic, in the promoter region, or not commonly encountered large deletion or duplication.

The specific Cystinosis symptoms vary greatly from one person to another based upon several factors including the age of onset and diagnosis of the disease. The symptoms of the three different forms of Cystinosis are as follows:
• Nephropathic Cystinosis: Growth failure, Fanconi syndrome, deterioration of the filtration function of the kidneys, etc.
• Intermediate Cystinosis: Also known as nephropathic juvenile cystinosis or adolescent cystinosis, this form of cystinosis is characterized by all of the signs and symptoms of nephropathic cystinosis described above. • Non-nephropathic Cystinosis: The disorder appears to affect only the eyes. Untreated individuals with non-nephropathic cystinosis eventually develop photophobia due to cystine crystal accumulation in the eyes.
| Types of Cystinosis | Symptoms | Onset |
|---|---|---|
| Nephropathic Cystinosis | Growth failure, Fanconi syndrome, deteriorated kidney filter function | Infancy |
| Intermediate Cystinosis | Symptoms of Nephropathic Cystinosis + muscle deterioration, blindness, inability to swallow | 12-15 years of age |
| Non-nephropathic Cystinosis | Photophobia, crystal accumulation in eyes | Above 20 years of age |
Cystinosis Market Insights and Epidemiology
As per DelveInsight, the total Cystinosis prevalent cases in the United States were found to be around one thousand cases in 2020, which might increase in the coming years. The majority of the cases of Cystinosis are accounted by Nephropathic Cystinosis, while the least cases are contributed by Ocular Cystinosis.
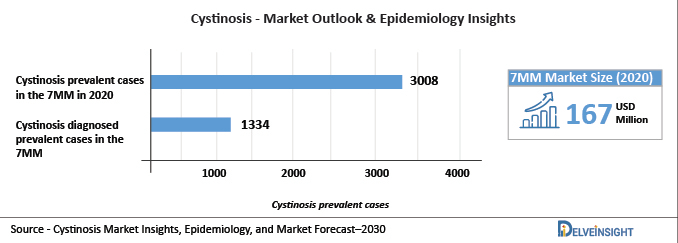
DelveInsight analysis indicates that total Cystinosis prevalent cases in the 7MM were found to be 3,008 in 2020, which might increase in 2030. These prevalent cases are expected to increase with a CAGR of 0.92% by 2030. Also, the total Cystinosis diagnosed prevalent cases in the 7MM were found to be 1,334 in 2020
Cystinosis market size in the 7MM is expected to rise from USD 167 million in 2020 and is expected to rise and cross more than USD 500 million in the upcoming years.
The increase in the Cystinosis market size is a direct outcome of the increasing prevalent population of Cystinosis cases, raising awareness, support and engagement with the community, as well as the introduction of innovative treatment options.
Cystinosis Treatment Therapy by Avrobio
Various Cystinosis therapies are approved for the two types – Nephropathic and Ocular Cystinosis. Pediatricians, kidney specialists (nephrologists), eye specialists (ophthalmologists), digestive disorder specialists (gastroenterologists), psychologists and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Cystinosis treatment includes cystine depleting therapy (Cystagon, Procysbi, Cystaran, Cystadrops, etc.); symptomatic therapies; renal transplantation, etc.
Due to the lack of investment by the pharmaceutical industry, together with the limited stability of cysteamine, very few drugs such as Procysbi, Cystadrops, Cystaran, and Cystagon are available in the market. As Procysbi was found to have fewer side effects than Cystagon, approval of Procysbi affected the market of Cystagon, both of these drugs are approved for nephropathic Cystinosis. The patent of the drug is also expected to expire between 2027 and 2036. The market size of Procysbi in 2020 was close to USD 130 million, whereas the market size of cystagon was approximately USD 1 million in 2020. It is also believed that Cystagon patients switched to Procysbi because of better safety profiles. There is a positive growth seen in the market size of Procysbi in 2030, whereas the market size of cystagon is expected to plummet down in 2030.
Similarly for Ocular Cystinosis, Cystaran was the first approved drug and had no competitor till 2019. Cystadrops entered the US market in 2020 and decreased the market captivity of Cystaran. As per Delveinsight’s estimates, Cystadrops is expected to achieve its peak sales by 2026. The former covers all the present patient pools falling under this category covering the market size of USD 1 million in 2020 which is expected to see a fall in the upcoming years. The latter in 2020 generated a market revenue of USD 1 million which is expected to surge in the years to come.
Cystinosis patients take dozens of pills a day on a strict schedule 24 hours a day, but oral cysteamine does not reach the cornea effectively and therefore does not remove cystine crystals from the cornea, thus patients should use all eye drops in addition to a few waking hours. Additionally, patients suffer from various side effects such as nausea and a pungent sulfur odour caused by the medication. Patients face a great challenge to adhere to the treatment regimen.
In order to overcome these challenges, Avrobio in collaboration with the University of California, San Diego (UCSD), is coming up with lentiviral-based gene therapy designed to potentially halt or reverse the progression of cystinosis with a single dose of the patient’s hematopoietic stem cells named AVR-RD-04 (CTNS-RD-04). This Cystinosis treatment therapy has received both Orphan and Fast Track Designation. In addition to this, gene therapy comes up with the hope of a one-time fix. The therapy doesn’t have to reach every cell for it to work, just enough to make enough protein that the cystine removes so it doesn’t build up. Rather than simply delivering a missing gene to cells, editing permanently changes a patient’s native DNA to insert a gene or turn off a gene that’s causing problems. This gene therapy contains a human gene for cystinosin designed to maximize the likelihood of sustained cystinosin production in hematopoietic stem cells and their progeny. It is manufactured from hematopoietic stem cells that are first extracted from the patient, modified to add the gene that encodes for cystinosin, and then infused into the patient.
Avrobio announced initial clinical data from the first three patients dosed in the investigator-sponsored Phase I/II trial of AVR-RD-04, the company’s investigational Cystinosis gene therapy. Since it’s a gene therapy—a one-time treatment—it has the potential to provide a permanent cure for Cystinosis disease. Hence it will be a major factor for attracting the patient pool. It is estimated that this therapy will give tough competition to emerging and other approved therapies and generate a major market share.
CAR T-cell therapies engineered using lentiviral vectors have demonstrated noteworthy clinical success in patients with B-cell malignancies leading to regulatory approval of the first genetically engineered cellular therapy using lentiviral vectors. Taking this into consideration, if approved AVR-RD04 will capture a large market size not only because of its better results but also because gene therapies are expensive. It is expected to cover the patient pool of nephropathic Cystinosis as well as Ocular Cystinosis.
Significant medical unmet needs still exist for patients with Cystinosis. Gene therapy represents a promising new therapeutic approach for the treatment of Cystinosis that may have the potential to address most of the complications associated with this disease, and the current clinical trials are positively anticipated to address the medical requirement in the patients.
Frequently Asked Questions (FAQs)
Renal allografts and medical therapy target the basic metabolic defect that has altered Cystinosis. It has improved the Cystinosis life expectancy drastically reaching beyond 50 years. Also, early diagnosis and appropriate therapy are very important too.
Kidney transplantation and Dialysis are substitute treatments for Cystinosis. They do not permanently cure the genetic condition but it is cured to some extent. However, the disease continues to affect every other organ in the body. Cystinosis causes growth failure as well as short stature.
Nephropathic Cystinosis is a genetic rare disease that appears in infants and children at a very young age. It is considered to be a life-long condition, but there are some treatments available, such as cysteamine therapy and kidney transplantation.
Ocular cystinosis is the benign, adult form of Cystinosis, a metabolic disease characterized by an accumulation of cystine crystals in the cornea and conjunctiva responsible for tearing and photophobia and associated with no other additional manifestations.
Downloads
Article in PDF
Recent Articles
- Gaucher disease Market Analysis: Rich Pipeline Drives the Market Ahead
- Novartis’ Canakinumab for NSCLC; Novartis’s Zolgensma Updates; Trodelvy Prospects in New Breast C...
- Biohaven’s Troriluzole Failure; Daiichi/ AZ’s Enhertu; Fujifilm & Manufacturing ...
- DelveInsight’s Infectious disorders based Gene Therapy Reports
- Merck’s KEYTRUDA as Adjuvant Therapy for RCC Patients; BMS Receives Positive CHMP Opinion for CAR...