Beta Thalassemia Treatment – Insights Into the Challenges & Solutions
Jan 21, 2022
Table of Contents
Beta Thalassemia (β-Thalassemia) is a blood disorder characterized by reduced synthesis of the hemoglobin subunit beta (hemoglobin beta chain) that results in microcytic hypochromic anemia, an abnormal peripheral blood smear with nucleated red blood cells, and reduced amounts of hemoglobin A (HbA) on hemoglobin analysis. General occurrence of severe and untreated Beta Thalassemia symptoms include severe anemia, with an Hb level of 3−6 g/dL, massive hepatosplenomegaly, severe growth retardation, and bony deformities. Beta Thalassemia treatment therapy involves regular blood transfusions, iron chelation, bone marrow transplant and novel gene therapy options. Surgery for spleen or gallbladder removal serves as the last curative treatment option.
People with Beta Thalassemia major or intermediate usually have a buildup of iron, either from the disease itself or from repeated blood transfusions. Excess iron can damage the heart, liver, and endocrine system. People with the minor form of Beta Thalassaemia have small red blood cells but no symptoms.
Downloads
Click Here To Get the Article in PDF
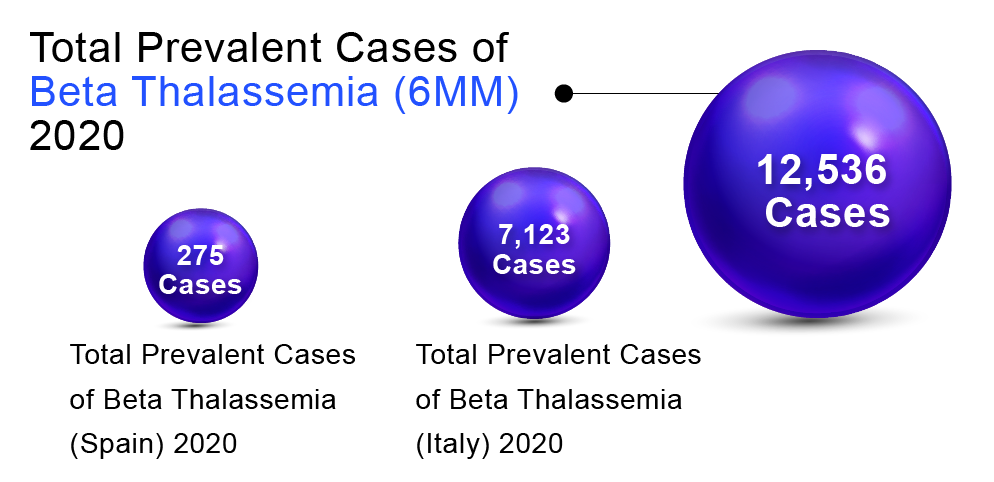
In order to determine Beta Thalassemia prevalent population in the 6MM (United States, Germany, France, Spain, Italy, and the United Kingdom), DelveInsight considered and compared data from various studies and organizational databases. We also collected different KOL views and expert opinions to cover the geographical differences in Beta Thalassemia epidemiology across the 6MM.
The total prevalent cases of Beta Thalassemia in the 6MM were found to be 12,536 in 2020. Italy accounted for the highest number of cases, followed by the United States, Germany, UK, France, and Spain. According to DelveInsight’s estimates, among the European countries, Italy had the highest number of prevalent cases of Beta Thalassemia (7,123) in 2020 and is expected to reach 7,058 by 2030.At the same time, Spain had the lowest number of prevalent cases (275)in 2020, which as per DelveInsight’s estimates, are expected to reach 280 cases by 2032.
“As per DelveInsight assessments, EU5 countries together accounted for 10,522 cases.”
Beta Thalassaemia Pathophysiology
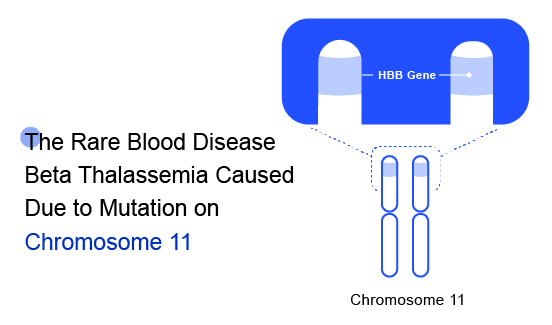
Beta Thalassaemia is an inherited blood disorder caused due to genetic modification on Chromosome 11. The major risk factors include “Family history of the disorder” and /or “Asian, Chinese, Mediterranean, or African American ethnicity.” These can become so severe that they may cause organ failure, which is when your organs stop working effectively. Beta Thalassaemia inheritance results from various mutations (over 200 disease-causing mutations have been identified) or, rarely, deletions of the beta-globin gene (HbB) on chromosome 11. These mutations are primarily ‘point mutations’ that affect transcriptional control, translation, and splicing of the HbB gene and gene product.
There are three main Beta Thalassaemia types including; Thalassaemia Major (variably referred to as “Cooley’s Anemia” and “Mediterranean Anemia), Thalassaemia Intermediate, and Thalassaemia Minor (also called “Beta Thalassemia carrier,” “Beta Thalassemia trait” or “heterozygous Beta Thalassemia”).
Disease associations and other organized programs for Beta Thalassemia patients are established in most regions. Some prominent ones include the Thalassemia Support Foundation, Thalassemia International Federation, and The Cooley’s Anemia Foundation, among others. All of these provide opportunities to map the patients and improve their access to innovative Beta Thalassemia treatment and outcomes. Soon, establishing country-wide registries in cooperation with these centers may also help tackle the disease further.
Transfusion-dependence is essential in distinguishing the various Thalassemia phenotypes and their severity. The diagnosis of Beta Thalassemia major entails a lifelong regular transfusion requirement for survival (TDT – Transfusion dependent thalassemia). Globally, ~100,000 patients currently receive regular transfusions for Beta Thalassemia. Blood transfusion exposes patients to several risks (blood-borne infection, alloimmunization, and iron overload), necessitating proper clinical practice guidelines for the management of TDT.
Severe Beta Thalassemia major patients require lifelong therapy to prevent and manage the clinical consequences of the disease. Long-term adherence to treatment is essential. Comprehensive management guidelines for both TDT and NTDT have been developed by the Thalassemia International Federation and are widely available. Current management strategies for TDT include blood transfusion, iron chelation, splenectomy (less common than in the past), and HSCT for a subset of patients.
The other medications used for Beta Thalassemia treatment include deferiprone (DFP), deferasirox (DFX), and desferrioxamine (DFO). Treatment with DFX and DFP is preferred due to their cost-effectiveness and ease of administration (oral). DFO, on the other hand, is administered subcutaneously, intravenously, or occasionally intramuscularly. The goal of Beta Thalassemia treatment is to maintain a hemoglobin level of 9–10.5 g/dL, which has been shown to promote normal growth, suppress bone marrow activity, and minimize iron accumulation.
Beta Thalassaemia Treatment Market Outlook
According to DelveInsight, the lion’s share of the Beta Thalassemia market by the end of the forecast period (2019–2032) is likely to be captured by the United States by reaching USD 745.1 million by 2032 from USD 74.9 million in 2019.
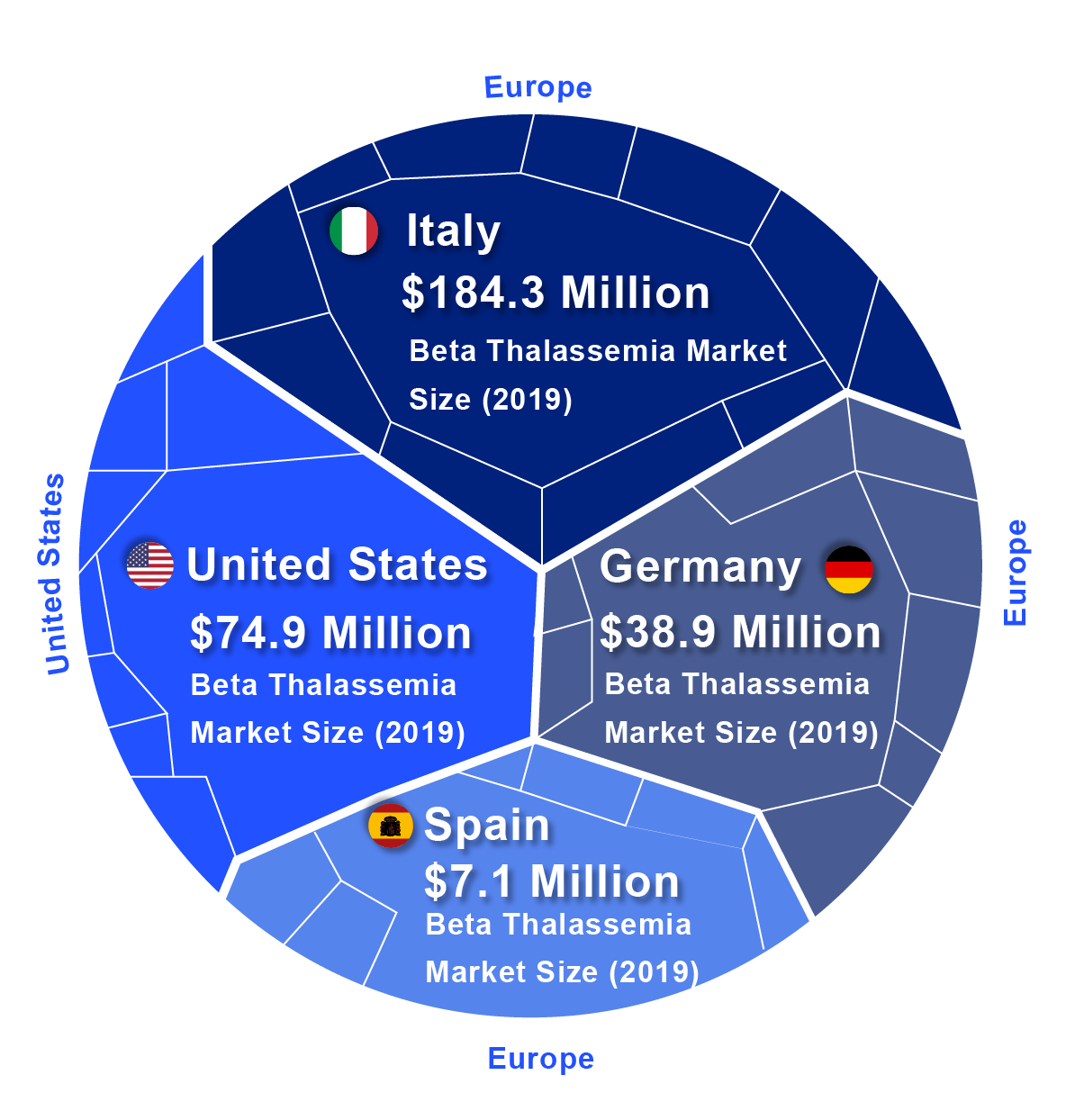
In 2019, among EU5, Italy accounted for the largest Beta Thalassemia market size (USD 184.3 million), followed by Germany with USD 38.9 million, whereas Spain accounted for the smallest (USD 7.1 million).
The future of the Beta Thalassemia treatment market looks promising with new and emerging therapies such as CTX001 by CRISPR Therapeutics/Vertex Pharmaceuticals, Mitapivat (AG-348) by Agios Pharmaceuticals, 9.4 IMR-687 by Imara Inc., among others.
As per DelveInsight Analysis, Anticipated United States #BetaThalassemia Market Size Will Reach Upto USD 745.1 million by 2032
Market Leaders in the Making
With a cornucopia of drugs in the Beta Thalassemia pipeline, the market size is projected to increase. Among the emerging therapies, the most promising ones include CTX001 – a Phase II/III Cell replacement therapy, Mitapivat – a Phase III Pyruvate Kinase stimulant, Zynteglo- Phase III Gene therapy (US), among others. Other promising therapies include IMR-687 (Imara Inc.), Sapablursen (Ionis Pharmaceuticals), DST-0509 (DisperSol Technology), SLN 124 (Silence Therapeutics), and many more.

Expected Roadblocks
At present, there remains a large degree of disparity in blood donation levels worldwide. Australia, Europe, and North America have the highest donation rates. In recent years, there has been a rise in donation rates, but the demand for transfusions in low-income countries exceeds the current supply. Around the globe, more than 22,500 patients with transfusion-dependent Beta Thalassemia die each year due to insufficient blood supply. Because of this, service providers are often required to seek out alternative blood sources, such as paid donors or family members.
However, international organizations such as WHO and the Red Cross discourage paid blood donation. Offering money may attract unhealthy donors, leading to anemia due to frequent donations, and drive altruistic donors away, resulting in fewer donors overall. Better awareness programs on blood donation may help improve the rates of blood donations.
Thechanging patterns of Thalassemia prevalence in different parts of the world are mainly affected by changes in the number of births of new patients. They are largely influenced by prevention programs, population migration, and improved survival of β‐thalassemia patients. Historically, β‐thalassemia is highly prevalent in the Mediterranean, Middle East, and Southeast Asia, and migration patterns have increased the prevalence of this hemoglobinopathy in North America and Northern European countries. Notably, the increasing prevalence in these countries has increased the burden on healthcare systems. As long as migration to these regions continues, for example, due to economic, social, or environmental reasons, the prevalence of β‐thalassemia will continue to increase until the point is reached where large‐scale preventive measures are implemented. The COVID‐19 pandemic has significantly diminished migration, but its long‐term consequences remain to be seen.
Another service essential for accurate diagnosis and monitoring of Beta Thalassemia patients is good laboratory support. TIF and other guidelines clearly state the need for diagnostic workup before the first transfusion, and they provide a timetable and a list of parameters required for patient monitoring throughout their life. Such parameters determine treatment decisions and must therefore be followed. Yet thousands of patients are treated according to a Hb and ferritin check performed at irregular intervals. Proper monitoring will give early warning of complications and has allowed lifesaving interventions. Universal standards for specialized tests such as Magnetic resonance imaging for the quantitation of iron load are still pending in many parts of the world.
Different Treatments For Beta Thalassemia
Iron is present in transfused blood, which is rarely eliminated by the body. On a typical blood transfusion schedule, patients with thalassemia collect iron at a rate of 0.3–0.6 mg/kg of body weight every day. At levels of 12–24 g of total body iron, excess iron becomes toxic and accumulates in the heart, liver, and endocrine system. Cardiomyopathy can be caused by too much iron, and untreated patients may die of heart failure before they reach the age of 20. Even in the absence of chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infections, iron excess raises the risk of liver fibrosis, cirrhosis, and hepatocellular cancer. Hypogonadism, growth retardation, delayed puberty, infertility, diabetes, and hypothyroidism are all possible side effects of iron overload. Excess iron must be removed to avoid consequences from iron overload, and ICT should begin within a year of starting frequent blood transfusions. ICT increases overall survival and lowers the incidence of iron overload consequences such as heart disease, short stature, and endocrine problems. In the United Kingdom, most patients now undergo ICT with either deferasirox or deferiprone monotherapy or combination ICT (typically subcutaneous desferrioxamine and deferiprone), which has significantly impacted how patients and others around them view and feel thalassemia.
Allogeneic hematopoietic stem cell transplantation (HSCT) is the only available curative treatment for Beta Thalassemia major (b-TM) patients. In recent years, the increased number of transplants has further upsurged with an increased incidence of Graft Versus Host Disease (GvHD). At the time of the survey, a higher incidence of comorbidities with GVHD was recorded as compared to those without it. Mortality in the first 10 years following HSCT was significantly higher than conventional treatment, with nearly 50% of the mortality being driven by acute GvHD as quoted in literature. Unfortunately, the incidence of acute GvHD following transplantation has not significantly improved over the decades, and hence lowering the rate of acute GvHD-related mortality remains an unmet goal.
Patients with transfusion-dependent Thalassemia must be managed with blood transfusions and iron chelation therapy for the rest of their lives. Iron overload complications can result in severe morbidity and mortality, and it is widely established that the chronic management of this disease places a significant financial and time burden on patients. Patients with TD-thalassemia had considerably greater healthcare resource utilization, prescription expenditures, and total costs than matched controls and were forced to contact their healthcare provider frequently, which hampered their quality of life and work productivity.
HAVE YOU READ? “How Gene therapy is changing the Beta-thalassemia Treatment outlook?”
The Road Ahead For Beta Thalassaemia
The futuristic approach for Beta Thalassemia treatment will depend on the increased availability of iron chelation, γ-globin induction, blood transfusion, allogeneic bone marrow transplantation, and gene therapy for Beta Thalassemia in developed as well as more in the under-developed and low-income countries. The next few years will be very critical for the rise of Beta Thalassemia treatment measures to be adopted, with enormous potential of gene transfer methods for Beta Thalassemia patients, in regard to both the safety and efficacy determinants.
As per DelveInsight estimates, the Beta Thalassemia market landscape looks promising with greater advances in diagnosis and treatments translating into a better quality of life and improved survival. The entry of the novel therapies necessitates collaboration among academia, life-sciences companies, funding, regulatory agencies, decision-makers, and caregivers to attain better healthcare.

Frequently Asked Questions
Beta Thalassemia is an inherited blood disorder that reduces the production of hemoglobin. A low level of hemoglobin usually leads to a lack of oxygen in many parts of the body.
Yes, Beta Thalassemia can be fatal with severe life-threatening symptoms and severe anemia, but with proper and regular treatments including blood transfusions, early deaths can be prevented.
Currently, the only complete cure is Bone Marrow Transplant for Beta Thalassemia, but it is possible for only a small proportion of patients.



