Fabry Disease – A Market Perspective On The Emerging Pipeline
Sep 04, 2023
Table of Contents
Fabry disease is a rare hereditary lysosomal storage disorder that is caused by mutation in the GLA gene located on the X chromosome. The defect leads to the deficiency of an enzyme called alpha-galactosidase A, responsible for the breakdown of globotriaosylceramide (Gb3). The accumulation of the fatty substance leads to systemic complications in multiple systems of the body including skin, eyes, heart, and kidneys. The condition presents with dermatological manifestation in its early stages but progresses to develop cardiovascular issues resulting in arrhythmias, heart failure, and attacks at a later point in time.
Fabry Disease: Epidemiology and Statistics
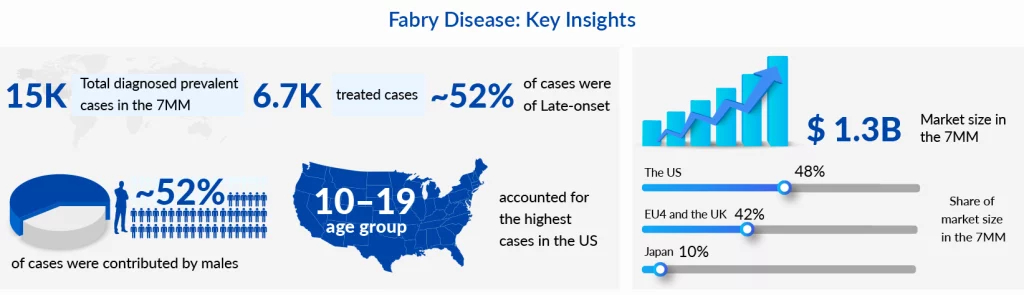
Fabry disease is the second most prevalent genetic metabolic storage disorder after Gaucher disease. Despite this, it has had severe diagnostic pitfalls due to its heterogeneity and diagnostic barriers. In 2022, there were more than 15,000 diagnosed Fabry disease cases in the 7MM, of which the US had the highest share with around 8,400 cases. The rise in newborn screening in developed countries, especially in the US, has led to a surge in the birth prevalence of the disease. However, there is no uniformity in the implementation of genetic screening in different states in the US which causes the Fabry disease prevalence to range from 1:6500 to 1:12000. The disease is known to affect both genders. It is estimated to have more number of patients with the milder late-onset phenotype than the more severe classic phenotype, at least in the 7MM.
Downloads
Article in PDF

Current Fabry Disease Treatment Market Outlook and Persisting Unmet Needs
The current Fabry disease treatment paradigm involves enzyme replacement therapies (ERT), along with oral chaperone therapy and adjunctive therapies. ERTs emerged with the launch of FABRAZYME (Sanofi) in 2001 in the EU, and subsequently in 2003, and 2007 in the US, and Japan, respectively. The brand became a standard of care in Fabry disease patients due to the availability of long-term safety and efficacy results. In 2018, JCR Pharmaceuticals introduced its biosimilar in Japan which offered a pricing advantage over the originator drug.
Replagal, another Enzyme therapy, was also introduced in the EU by Shire in 2001 and in Japan by Dainippon Sumitomo Pharma in 2007. The drug contains the active substance agalsidase alpha and is administered through the parenteral route of administration. Though Shire filed a BLA for FDA approval in 2009, the drug could not acquire approval due to the additional data requirements in the US.
Over a decade after the last approval of any therapy for Fabry disease, Amicus was able to introduce its oral chaperone therapy GALAFOLD in the European market in 2016. Subsequently, in the year 2018, the drug was approved in the US and Japan.
Though these Fabry disease therapies have proven to be beneficial in the management of Fabry disease patients, significant challenges and unmet needs still exist. The foremost concern that ERTs pose is due to their repeated parenteral infusions that cause patient inconvenience and reduced compliance. The frequent dosing is due to the short half-life of these replacement therapies. Additionally, there are patients who lose access through peripheral intravenous routes and need central venous catheter due to multiple parenteral administrations. Moreover, some of the patients who are treated with ERT develop immune responses through anti-drug antibodies (ADAs) targeting the recombinant enzyme and causing a reduction in efficacy. Enzyme replacement therapies might slow down the progression of the complications in the patients but renal or cardiac concerns still develop in most of the cases. On the other hand, chaperone therapies have more merits in terms of their oral route of administration vs. the burden of biweekly infusions. However, the drugs are available only for individuals with specific amenable mutations, and around 50-65% of Fabry disease patients may not be eligible for them. This presents an additional limitation with such therapies restricting their access to a few patients. Lastly, the existing therapies do not treat the underlying causative factor and may only serve as symptomatic solutions for Fabry patients.
Emerging Therapies and Their Place in Evolving Fabry Disease Treatment Approach
The growing number of Fabry disease patients is posing a pressing demand for better Fabry disease treatment options with minimum adverse effects. Pharmaceutical companies have been working on addressing the unmet needs of existing therapies through the application of novel mechanisms.
ELFABRIO (PRX-102), developed by Chiesi and Protalix, was approved in 2023 in the US and the EU. The drug is a PEGylated enzyme replacement therapy (recombinant ERT) having a longer half-life than the conventional ERT. Due to its comparatively greater stability in the blood, the dosing frequency is once a month. Furthermore, the drug, in its clinical trial, exhibited a significant improvement in kidney functions making it an attractive candidate to gain major patient share in the Fabry disease treatment market. The drug eliminates a major unmet need of the traditional ERT which is the development of ADAs and maintains its efficacy in such cases. Around 40% of ERT-treated individuals have sub-optimal efficacy due to generation of antibodies. The drug is anticipated to penetrate the market well, targeting both treatment-naive patients as well as those facing limited effectiveness with existing treatments. It is assumed to peak with a 15% patient share reaching ~USD 500 million by 2032. It should be noted that the drug has not been approved in Japan but Chiesi has its trial ongoing in the region and could get its approval by 2027.
Another important emerging Fabry disease therapy that is likely to hit the market by 2025 in the US is Isaralgagene civaparvovec (ST 920) which is being developed by Sangamo Therapeutics. The gene therapy would offer the advantage of a one-time administration. The drug is being tested in Phase II and targets to obviate the requirement of ERT infusions. Additionally, the drug is likely to have low immunogenicity which would cause minimal need for immunomodulating agents. Despite multiple factors bolstering its prospects in the emerging Fabry disease treatment market, the existing gene therapies allude toward low penetration in the rare disease markets due to cost implications and lower physician and patient preferences. The gene transference treatment could attain a peak patient share of 2% in the US with even less in the European and Japanese Fabry disease treatment market. The drug could attain its peak within 7 years of launch with sales of ~USD 350 million by 2032 in the 7MM.
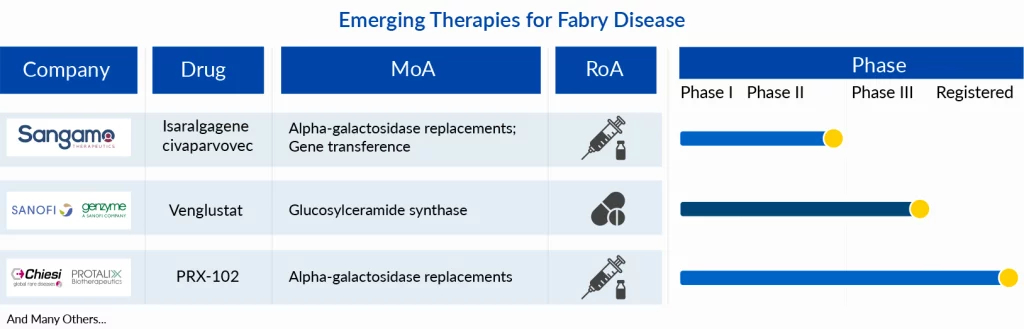
Lastly, a drug that leverages a substrate reduction mechanism is also underway. Sanofi is investigating an oral therapy in Phase III which could target the Fabry disease patient pool with concomitant complications of ventricular hypertrophy or neuropathic and abdominal pain. The small molecule has shown signs of reduction in Gb3 in its proof of concept trial and its efficacy is yet to be determined in pre-marketing clinical trials.
Analyst Assessment
As per the estimations by analysts at Delveinsight, the market in the 7MM is likely to cross USD 2.4 billion by 2032 with 35% contribution from conventional enzyme replacement therapies. All in all, the Fabry disease market conditions are conducive to the growth of novel drugs and the trend will continue unperturbed for a few years in the absence of any major disruptions such as biosimilars or other factors.
FAQs
Fabry Disease is an inherited lysosomal storage disease caused by a nonfunctional or partially functional enzyme called alpha-galactosidase A (α-Gal A). Decreased activity of α-Gal A in lysosomes results in the accumulation of enzyme substrates (Gb3 and lyso-Gb3) which cause cellular damage in tissues throughout the body.
Currently, there is no permanent cure for Fabry Disease. But, recent developments in the Fabry Disease treatment scenario are leading to a symptomatic cure, relieving the condition.
Fabry Disease is rare, inherited, and a serious genetic disorder. It is a lipid storage disorder that can cause long-term difficulties in the kidneys, heart, and nervous system. Fabry Disease can be understood as a terminal illness as well.
Fabry Disease involves many potential life-threatening complications such as progressive kidney damage, heart attack, and stroke. It can be fatal.

Downloads
Article in PDF



