5 Promising Pompe Disease Drugs in Early-stage Development
Nov 01, 2024
Table of Contents
Imagine a disease that stealthily incapacitates the heart, liver, and skeletal muscles—slowly breaking them down due to the absence of a crucial protein. That’s the story of Pompe disease, also known as glycogen storage disease type II (GSDII). Caused by a gene mutation that blocks the body’s ability to break down glycogen, Pompe leads to a harmful buildup of sugar, disrupting the architecture of vital organs.
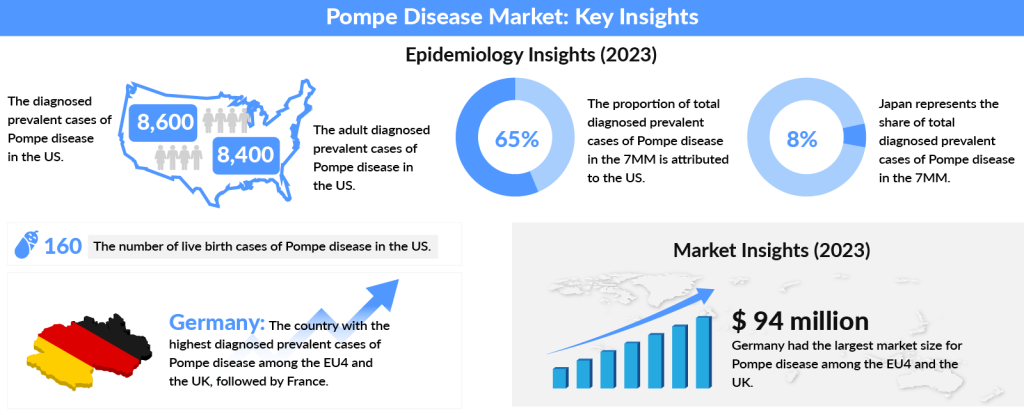
On a global scale, Pompe disease affects approximately 2.0 cases per 100,000 live births, making it a rare yet significant condition. The late-onset form tends to be more common than the early neonatal type. Across the 7 major markets (7MM)—the US, EU4 (Germany, France, Spain, Italy), the UK, and Japan—the prevalence varies, but the numbers are rising. In 2023, the US saw the highest cases, accounting for 65% of diagnosed cases, with roughly 8,400 adult patients. With projections suggesting growth in these numbers by 2034, the need for breakthroughs in treatment has never been more critical.
Downloads
Article in PDF
Recent Articles
- Exploring the Nonalcoholic Steatohepatitis (NASH) Landscape
- In vivo monitoring of T cell–dendritic cell interactions by intercellular enzymatic labelling
- Meet DelveInsight in BioEurope '16
- Global Hemophilia Market
- Ethicon’s ETHIZIA Hemostatic Sealing Patch; FDA Approves Medtronic’s Minimally Invasive Device to...
Discover everything you need to know about Pompe disease in our latest blog!
In the battle against Pompe disease, hope is rising like the sun breaking through the clouds! This rare genetic disorder, caused by a deficiency of the enzyme acid alpha-glucosidase, leads to glycogen buildup in the body, wreaking havoc on muscles and organs. Currently, the standard of care involves enzyme replacement therapies (ERT) like POMBILITI plus OPFOLDA (Amicus Therapeutics), NEXVIAZYME (Sanofi), and LUMIZYME/MYOZYME (Sanofi). While these Pompe disease treatments enhance survival by tackling cardiac issues, they fall short in addressing skeletal muscle and central nervous system complications. Early newborn screening for Pompe disease is essential for timely intervention, which can prevent irreversible muscle damage and unlock the greatest clinical benefits.
Curious about the latest treatments for Pompe disease? Check out our newest blog for all the details right here!
A Glimpse into the Future: Promising Innovations in Pompe Disease Treatments
The pipeline for Pompe Disease drugs is limited, emphasizing the urgent need for new Pompe Disease treatments. As we cast our gaze toward the future, ongoing clinical trials for investigational innovative medicines shine a beacon of hope, promising to expand treatment options and inch closer to a Pompe disease cure. The landscape of Pompe Disease treatments and Pompe Disease drugs is evolving, highlighting the critical role of groundbreaking approaches in tackling this challenging condition.
Let’s take a deep dive into the 5 most promising therapies in the Pompe disease drug pipeline
Actus Therapeutics’ ACTUS-101
Phase – I/II
ACTUS-101 is an adeno-associated virus (AAV) gene therapy designed to deliver a functional GAA gene directly into muscle cells, addressing the underlying deficiency that causes Pompe disease. ACTUS-101 is currently undergoing a Phase I/II Pompe disease clinical trial that began dosing patients in January 2019. This trial is designed to evaluate the safety, bioactivity, and immune responses to a single infusion of ACTUS-101, with the primary focus being on treatment-emergent adverse events (TEAEs). Secondary endpoints include muscle AAV bioactivity, serum GAA bioactivity, and functional assessments like the six-minute walk test. While the trial remains active, it is not currently recruiting, and updates on patient outcomes have been sparse since the initial dosing started.
If successful, ACTUS-101 could replace traditional ERT administered bi-weekly, offering a more convenient treatment option for patients. Further data from ongoing trials will be critical in determining its efficacy and safety profile, contributing to the broader landscape of Pompe Disease treatment.
Valerion Therapeutics’ VAL-1221
Phase – I/II
VAL-1221 is an investigational intravenous treatment for Pompe disease, aimed at enhancing muscle strength and functionality in individuals affected by this rare genetic disorder. Originally developed by Valerion Therapeutics, the drug has now been transferred to Parasail for continued clinical development. Pompe disease results from a genetic mutation leading to the deficiency of the enzyme acid alpha-glucosidase (GAA), causing glycogen to accumulate primarily in muscle tissues. This accumulation disrupts cellular functions, leading to muscle weakness and potential respiratory complications.
Unlike traditional enzyme replacement therapies (ERT), which primarily target glycogen stored in lysosomes, VAL-1221 addresses both lysosomal and extra-lysosomal glycogen buildup. It is a fusion protein combining Valerion’s proprietary 3E10 delivery antibody with recombinant human GAA, allowing for broader delivery of the enzyme throughout the body. Administered as an intravenous infusion every other week, VAL-1221 has been tested in varying doses of 3 mg/kg, 10 mg/kg, and 30 mg/kg, aiming to provide a more effective treatment option for those suffering from Pompe disease. The drug candidate is currently in Phase I/II stage of clinical development by Valerion Therapeutics for the treatment of late-onset Pompe disease.
Amicus Therapeutics’ AT-GAA
Phase I/II
AT-GAA is an investigational two-part therapy combining cipaglucosidase alfa (ATB200), a novel recombinant human acid alpha-glucosidase (rhGAA) enzyme featuring optimized carbohydrate structures, notably bis-phosphorylated mannose-6 phosphate (bis-M6P) glycans, to improve cellular uptake, with miglustat (AT2221), which stabilizes cipaglucosidase alfa. Preclinical studies have linked AT-GAA to higher levels of the mature lysosomal form of GAA, reduced glycogen in muscle, correction of autophagic defects, and improvements in muscle strength. Currently, Amicus Therapeutics is evaluating AT-GAA in phase I/II clinical trials for Pompe disease.
Astellas Pharma’s AT845
Phase – I/II
Astellas Pharma’s AT845, an investigational adeno-associated virus (AAV) vector-based gene replacement therapy for treating late-onset Pompe disease (LOPD), has shown promising efficacy in interim data from the Phase I/II FORTIS clinical trial (NCT04174105). The data, presented at the WORLDSymposium 2023 in Orlando, Florida, indicated that AT845 could provide a new avenue for managing Pompe disease. The FORTIS trial, which began in 2022, assesses the therapy’s safety, tolerability, and exploratory efficacy.
In Sept 2022, It was revealed that four participants received intravenous infusions of AT845. Three of them were able to discontinue enzyme replacement therapy (ERT) and maintained stable functional outcomes off ERT for periods ranging from 19 to 51 weeks. Most treatment-emergent adverse events (TEAEs) were mild and resolved without significant issues. However, the trial faced a setback when a participant experienced grade 2 peripheral sensory polyneuropathy, leading to an FDA clinical hold in June 2022. Thankfully, this hold was lifted in January 2023, allowing Astellas to continue its groundbreaking work in advancing AT845 as a potential game-changer in LOPD treatment.
Astellas aims to continue advancing AT845 through ongoing evaluations. If successful, it may provide a novel treatment option that enhances patient quality of life by delivering a functional GAA gene directly to muscle tissues. This comprehensive overview highlights each drug’s development status, clinical trial details, significant results, and future prospects in treating Pompe disease as of September 2024.
GeneCradle Pharmaceutical’s GC301
Phase – I/II
The clinical exploration of GC301, a gene therapy drug developed by Beijing GeneCradle Pharmaceutical, is focused on treating infantile-onset Pompe disease (IOPD) using an AAV vector. This vector expresses a codon-optimized human acid alpha-glucosidase (GAA) gene, offering a promising therapeutic strategy for patients under six months old diagnosed with IOPD. The therapy aims to replace the defective GAA gene, allowing for functional enzyme production to reduce glycogen accumulation in cells.
Participants in the trial will receive one of two doses of the GC301 AAV vector via intravenous administration. The primary endpoints include measuring the number of participants with adverse events, dose-limiting toxicity, and changes in upright forced vital capacity (FVC), a marker of respiratory health. Secondary outcomes involve tracking the 6-minute walk test results, muscle function, quality of life, and immune responses to both the virus and the GAA protein.
In addition, researchers will analyze GAA enzyme activity in muscles and blood, alongside the amount of glycogen stored in muscle tissue. This multicenter, open-label study aims to enroll 33 patients in China, focusing on late-onset Pompe disease (LOPD) cases as well. Eligible participants must have an FVC of at least 30% and be able to walk a minimum of 40 meters. The trial, which began dosing participants on April 19, 2024, is projected to run until December 2026.
The gene therapy, GC301, offers a cutting-edge approach by utilizing a virus to deliver a functional GAA gene. This trial highlights China’s pioneering efforts in gene therapy development, particularly for a rare disorder like Pompe disease. Currently, GC301 remains in Phase I/II, but the drug’s approval by the National Medical Products Administration (NMPA) and the clinical trial ethics committee underscores the hope it brings for the future of Pompe Disease treatment in IOPD. However, the study is confined to China for now, with global eyes on its outcomes.
What’s the Future of Pompe Disease Treatment?
As the landscape of Pompe disease therapies evolves, we stand at the precipice of groundbreaking advancements that promise to redefine the treatment journey for patients and families grappling with this rare disorder. With innovative late-stage therapies like ACTUS-101, AT845, and GC301 making strides in clinical trials, the horizon glows with the potential for novel treatments that could reshape the lives of those affected by Pompe disease. These therapies not only aim to address the underlying genetic causes but also offer hope for improved quality of life, moving beyond the limitations of existing enzyme replacement therapies.
Discover the causes, symptoms, diagnosis, and treatment landscape of Pompe disease, including current therapies and emerging drugs. Dive in for a comprehensive overview!
The journey towards a Pompe disease treatment is not just a medical challenge but a shared vision that ignites hope in the hearts of many. As researchers, clinicians, and advocates rally together, the future looks bright. Each trial, each data point, and each breakthrough brings us closer to transforming Pompe disease from a once-daunting diagnosis into a manageable condition. The promise of these cutting-edge therapies is not just a testament to scientific ingenuity; it’s a beacon of hope for patients and families, illuminating the path to a healthier, brighter tomorrow.
With advancements in treatment for Pompe disease, we are not only improving outcomes but also positively impacting Pompe disease life expectancy with treatment. As we continue to follow the developments in this field, let us remain optimistic and engaged, championing every step forward in the relentless pursuit of a cure for Pompe disease.

Downloads
Article in PDF
Recent Articles
- Alzeimer’s drug; Guardant Health receives approval; First Patient Dosed in Phase 1 Clinical Trial...
- The Snippet: The Success of Cannabidiol
- Allergic Conjunctivitis (AC)- Market Insights, Epidemiology and Market Forecast- 2027
- FDA Expands SOLIRIS for Pediatric Myasthenia Gravis; vTv’s Cadisegliatin Program Resumes as FDA L...
- Eli Lilly’s COVID-19 Antibody trial; Approval for Oriahnn; Gilead’s Remdesivir Clinic...



