The Changing Face of Mucopolysaccharidosis Type I Treatment: Enzyme Replacement, Gene Therapy, and Beyond
Mar 24, 2025
Table of Contents
Mucopolysaccharidosis Type I (MPS I) is a rare genetic disorder caused by a deficiency of the enzyme α-L-iduronidase, leading to the buildup of complex sugars in the body. This progressive condition can impact multiple organs, causing skeletal abnormalities, vision, and hearing loss, and even cognitive decline.
There are three variants of MPS I, which differ widely in their severity. The severity of the condition depends on how much alpha-L-iduronidase (IDUA) activity is present. Hurler syndrome is the most severe, Scheie syndrome the mildest, and Hurler-Scheie syndrome gives an intermediate phenotype.
Severe MPS I is more common than attenuated MPS I. Children with severe MPS I begin showing symptoms before the age of one, with intellectual disabilities presenting by the age of three, and passing away within the first ten years of life. Approximately 60% of children born with MPS I have the most severe subtype and rarely live past the age of 10 when untreated.
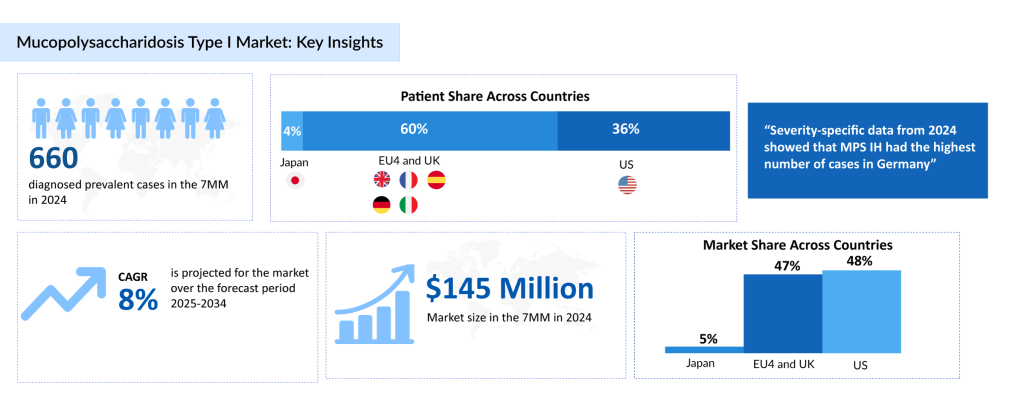
According to DelveInsight’s estimates, in 2024, there were approximately 660 diagnosed prevalent cases of MPS I in the 7MM. Of these, the US accounted for the second highest diagnosed prevalent cases of MPS I which are expected to increase by 2034 at a CAGR of 0.6% over the forecast period from 2025 to 2034.
Despite its challenges, breakthroughs in enzyme replacement therapy and stem cell transplants are giving new hope to patients.
ALDURAZYME—The Only Approved Drug for MPS I Treatment
Management of MPS I depends on the severity of the disease, with different approaches for severe (MPS IH) and attenuated (MPS IH/S, MPS IS) forms. Hematopoietic stem cell transplant (HSCT) is the standard treatment for severe MPS I, especially in children under two, and may also be considered for attenuated cases. HSCT allows donor-derived cells to produce IDUA, which can cross the blood-brain barrier (BBB) and reduce central nervous system (CNS) involvement. It helps slow disease progression, preserve cognitive function, and improve survival, but it has a limited impact on skeletal issues and carries transplant-related risks.
Enzyme replacement therapy (ERT) with laronidase primarily targets non-CNS symptoms, improving liver and spleen size, respiratory function, mobility, and joint flexibility, with better outcomes when started early. However, ERT has limited CNS benefits due to poor BBB penetration, and immune responses can reduce its effectiveness. Research indicates that HSCT may help reduce these immune reactions, potentially enhancing overall treatment efficacy.
Despite its benefits, HSCT is generally limited to severe cases due to the risk of complications, including morbidity and mortality. Early diagnosis is essential, as treatment success depends heavily on the disease stage at the time of initiation. A combination of ERT and HSCT is emerging as a promising strategy, where short-term ERT before HSCT helps stabilize patients, reduce glycosaminoglycan accumulation, and improve engraftment success, leading to better long-term outcomes.
Currently, BioMarin Pharmaceutical/Sanofi’s ALDURAZYME (laronidase) is the only approved ERT for MPS I. The treatment with ALDURAZYME has been shown to improve walking capacity and pulmonary function. It is a polymorphic variant of the human enzyme a-L-iduronidase produced by recombinant DNA technology.
ALDURAZYME uptake by cells into lysosomes is mediated by the mannose-6-phosphate (M6P)-terminated oligosaccharide chains of laronidase binding to specific M6P receptors. ALDURAZYME provides an exogenous enzyme for uptake into lysosomes and increases the catabolism of GAG. It was first approved in the US in April 2003, followed by approvals in the EU and UK (June 2003) and Japan (2006).
Despite the patent expiration, no biosimilar or alternative biologic has been approved yet, highlighting a significant gap in treatment options for MPS I. The need for new treatments remains high, as existing therapies have certain limitations, including safety concerns such as a boxed warning for ALDURAZYME.
Promising Candidates in the MPS I Treatment Pipeline
The pipeline of MPS I is not robust, with only a few emerging drugs currently in development. Key players such as Kyowa Kirin and Orchard Therapeutics (developing OTL-203, a gene therapy targeting the underlying enzyme deficiency), JCR Pharmaceuticals (advancing JR-171, a blood-brain barrier-penetrating enzyme replacement therapy), Immusoft (working on ISP-001, a cell-based platform for sustained enzyme production), and REGENXBIO (developing RGX-111, a gene therapy designed to deliver a functional copy of the IDUA gene) are leading the charge in advancing treatment options. These innovative therapies aim to improve central nervous system involvement and overall patient outcomes, addressing significant unmet needs in the MPS I community.

With the introduction of new MPS I therapies in clinical development and a rise in disease awareness and diagnosis, the MPS I market is expected to grow steadily during the forecast period (2025–2034). The increasing focus on gene therapy, enzyme delivery across the blood-brain barrier, and more targeted approaches reflect a shift toward improving long-term outcomes and reducing the disease burden. As regulatory approvals and clinical success stories emerge, the competitive landscape for MPS I treatments is poised for significant transformation, offering renewed hope for patients and caregivers.
Key Developments in the MPS I Therapeutic Segment
- In March 2025, REGENXBIO Inc. finalized its previously announced strategic partnership with Nippon Shinyaku. Under the agreement, REGENXBIO and Nippon Shinyaku will collaborate to develop and commercialize RGX-111 for Mucopolysaccharidosis I (MPS I), also known as Hurler syndrome, in the United States and Asia.
- In February 2025, at the 21st Annual WORLDSymposium, Orchard Therapeutics presented updated findings from a proof-of-concept study of OTL-203, an investigational gene therapy for the Hurler subtype of mucopolysaccharidosis type I (MPS-IH), during an encore oral presentation. The presentation summarized key neurological, skeletal, and other clinical outcomes observed in the study.
- In January 2025, Immusoft announced positive results from the first engineered B Cell (ISP-001) in a human clinical trial, to be presented at the WORLDSymposium.
- In October 2024, Orchard Therapeutics announced two poster presentations at the European Society of Gene and Cell Therapy (ESGCT) 31st Annual Congress on OTL-203.
- In September 2024, JCR Pharmaceuticals presented a poster on JR-171 at the Study of Inborn Errors of Metabolism (SSIEM) Annual Symposium 2024, held in Porto, Portugal.
- In May 2024, Orchard Therapeutics announced oral and poster presentations at the American Society of Gene & Cell Therapy (ASGCT) 2024 on OTL-203 clinical outcomes from a proof-of-concept (PoC) study in the Hurler subtype of MPS I.
- In February 2024, Orchard Therapeutics reported the first patient’s randomization in the registrational trial of OTL-203 for MPS IH, marking a key milestone in the therapy’s clinical development.
Future of the MPS I Treatment Market Looks Bright
The future of MPS I treatment is poised for significant advancements, driven by ongoing research and innovative therapies. According to DelveInsight’s analysis, the MPS I market in the 7MM was valued at approximately USD 145 million in 2024. Over the forecast period from 2025 to 2034, this market is projected to grow at a CAGR of 8.0%.
Current approaches, including enzyme replacement therapy, hematopoietic stem cell transplantation, and supportive care, remain essential. However, emerging treatments have the potential to address unmet needs, improve effectiveness, and enhance outcomes.
As research progresses, integrating novel strategies will lead to a more comprehensive and effective approach to managing MPS I in the future. Emerging therapies, such as stem cell gene therapy, IDUA Gene therapy, and Recombinant DNA offer novel approaches.
Additionally, increased regulatory support and incentives for rare diseases are also accelerating research and development in the MPS I space. The rise in newborn screening programs is leading to earlier diagnosis, which improves the chances of successful intervention and better long-term outcomes. Moreover, the growing focus on patient-centric care and personalized medicine is driving the development of tailored therapies, enhancing both efficacy and patient experience.
As clinical trials for novel therapies show promising results and more MPS I treatment options reach the market, the MPS I market is expected to expand rapidly, offering hope to patients and families affected by this challenging condition.




