Advances in Pompe Disease Treatment: From ERT to New Therapies
Sep 30, 2024
Pompe disease, an inherited rare disease, is often fatal and incapacitates the heart and skeletal muscles. The absence of an essential protein due to the mutation in a gene encoding GAA, leads to a complex buildup of sugar, hence, damaging the heart, liver, and other organs. The mutated gene hampers the lysosomal-mediated degradation of glycogen resulting in its intralysosomal accumulation and disrupting the tissue architecture. This is why Pompe disease is also known as glycogen storage disease type II (GSDII).
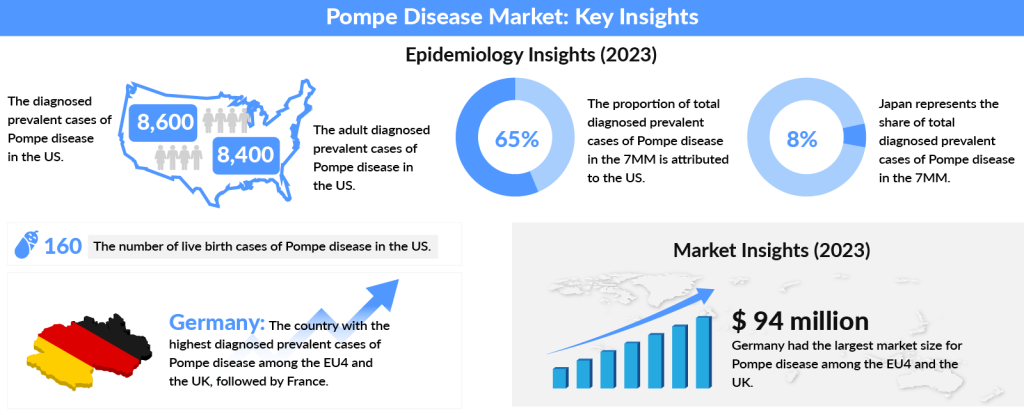
The worldwide Pompe disease incidence is 1 in 40,000 live births with the late-onset form of the disease generally being more common than the early neonatal form, as estimated by Clinical Therapeutics- Genetics and Epidemiology of Pompe disease. If we look at the Pompe disease prevalence over the years, the US National Library of Medicine National Institutes of Health, reported it to be approximately 1 in 28,000 in the United States.
In 2023, the US accounted for nearly 65% of diagnosed Pompe disease cases across the 7MM, with numbers projected to increase by 2034. The US had around 8,400 adult cases, while Spain recorded the fewest. Among the EU4 and the UK, Germany had the highest number of diagnosed cases, followed by France. Japan represented about 8% of the total diagnosed cases in the 7MM that year.
Downloads
Click Here To Get the Article in PDF
Recent Articles
- Assessment of Key Products that Got FDA Approval in Second Half (H2) of 2021
- 5 Promising Pompe Disease Drugs in Early-stage Development
- Pompe Disease: Causes, Symptoms, Diagnosis, Pathology, Current Marketed and Emerging Drugs in the...
- Immix Bio’s NXC-201 Gets FDA RMAT for AL Amyloidosis; Biodexa’s eRapa Wins Fast Track for Familia...
- Fourth FDA Approval for AbbVie’s Vraylar; FDA Approves Ferring’s Adstiladrin for NMIBC; Merck and...
Pompe Disease Therapeutic Market
As already mentioned above, the cause of Pompe disease is a lack of or insufficient amounts of an enzyme called acid alpha-glucosidase (GAA). The protein helps break down glycogen, which is required to produce energy. At present, there exists no cure for Pompe disease. However, different treatment options are available to relieve the Pompe disease symptoms. For instance, many patients used to undertake Supportive Therapy to tackle respiratory and cardiac problems, physical disability, and difficulties in swallowing before the arrival of Enzyme replacement therapy (ERT). ERT helped to prompt a new era in Pompe disease treatment. This was the first disease-specific treatment that was able to halt the progression of Pompe disease.
The Pompe disease therapeutic market features several FDA-approved drugs, each playing a crucial role in managing this rare genetic disorder.
MYOZYME (alglucosidase alfa) was the first drug approved by the FDA on April 28, 2006, for treating Pompe disease. It is an enzyme replacement therapy (ERT) specifically designed to treat infantile-onset Pompe disease by providing the human enzyme acid alpha-glucosidase, which helps break down glycogen. People with Pompe disease lack sufficient levels of this enzyme, leading to severe health issues.
LUMIZYME (alglucosidase alfa), approved by the FDA on May 25, 2010, for late-onset Pompe disease, is another ERT. It contains a naturally occurring enzyme that helps replace the missing one in patients with Pompe disease. Initially approved for individuals aged 8 and older, it was later extended in 2014 to treat Pompe patients of all ages.
NEXVIAZYME (avalglucosidase alfa-ngpt) received FDA approval on August 6, 2021, for treating patients aged one year and older with late-onset Pompe disease. This ERT targets the mannose-6-phosphate (M6P) receptor, essential for cellular uptake, and has shown improvements in respiratory function and walking distance in clinical trials.
POMBILITI (cipaglucosidase alfa-atga), a recently FDA-approved treatment as of September 28, 2023, is used alongside OPFOLDA (miglustat) capsules. This two-component therapy is for adults with late-onset Pompe disease who weigh at least 40 kg and are not responding to their current ERT. POMBILITI replaces the missing enzyme acid alpha-glucosidase, while OPFOLDA acts as an enzyme stabilizer, ensuring efficient glycogen breakdown.
Each of these therapies provides targeted treatments aimed at replacing or stabilizing the missing enzyme responsible for managing glycogen levels in patients with Pompe disease, helping improve their quality of life.
The Pompe disease therapeutic area is seeing rapid advancements as pharmaceutical companies race to develop innovative Pompe disease therapies that could revolutionize treatment and potentially improve Pompe disease life expectancy. Leading companies such as Amicus Therapeutics, Actus Therapeutics, Genzyme, Sanofi, Valerion Therapeutics, Astellas Therapeutics, Roche, Lacerta Therapeutics, and others are driving the Pompe disease pipeline forward. Their work focuses on developing new Pompe disease drugs that aim to address the root cause of the disorder and provide better outcomes for patients.

One of the most promising Pompe disease medicines in development is AT-GAA by Amicus Therapeutics. This innovative therapy, which is in Phase III trials, combines ATB200, a recombinant human acid alpha-glucosidase enzyme, with AT2221, a pharmacological chaperone that improves the enzyme’s targeting and uptake by cells. The recent positive results from the Phase III OLE study show that AT-GAA could significantly alter the Pompe disease treatment landscape, offering hope for improved outcomes and possibly extending Pompe disease life expectancy.
In addition to enzyme replacement therapies (ERT) like AT-GAA, Actus Therapeutics is exploring gene therapy as a long-term solution for Pompe disease with its candidate ACTUS-101. This Pompe disease treatment is an adeno-associated virus (AAV) gene therapy designed to increase the production of the GAA enzyme by targeting the liver. With the first patient dosed in its Phase I/II trial, ACTUS-101 could bring a new dimension to Pompe disease therapies by addressing the genetic root of the condition.
The future of the Pompe disease therapeutic area is bright with additional companies, such as Spark Therapeutics with SPK-3006 and Astellas Pharma with AT845, contributing to the expanding Pompe disease pipeline. These emerging Pompe disease drugs are expected to improve patients’ quality of life and offer more effective treatment options.
Despite the innovations, the high Pompe disease drugs price remains a challenge. The cost of ERT and new gene therapies continues to be a significant concern, but the value they provide in terms of improved health outcomes and extended life expectancy may justify their high costs. As more Pompe disease drugs uses are explored and therapies are approved, the Pompe disease drugs price is expected to remain high due to the complexity of developing and producing these life-saving medicines.
The journey of Pompe disease therapies has seen remarkable advancements, from supportive care to cutting-edge ERT like MYOZYME, LUMIZYME, NEXVIAZYME, and the recently approved POMBILITI in combination with OPFOLDA. These breakthroughs have not only transformed the Pompe disease therapeutic area but also offered renewed hope to patients and their families. While challenges such as the Pompe disease drugs price remain, the growing understanding of the disease, alongside innovative approaches like gene therapy and advanced chaperone therapies, are paving the way for a brighter future in Pompe disease treatment. The continuous expansion of the Pompe disease pipeline promises to extend Pompe disease life expectancy, improve patient outcomes, and provide more effective, life-changing Pompe disease drugs in the years to come.

Downloads
Article in PDF
Recent Articles
- 5 Promising Pompe Disease Drugs in Early-stage Development
- Fourth FDA Approval for AbbVie’s Vraylar; FDA Approves Ferring’s Adstiladrin for NMIBC; Merck and...
- BeiGene’s Brukinsa Approval; FDA Approval to Seagen’s TUKYSA; NICE Recommends Alnylam’s Amvuttra;...
- The Transforming Pompe Disease Treatment Landscape in Emerging Markets
- Avrobio taps Magenta’s ADC; Eli Lilly ramps up; FDA postpones decision; CSL gears up to fight pan...



