Vertex/CRISPR’s Gene-editing Therapy exa-cel: Inch Ahead of Rival
Apr 14, 2023
Vertex Pharma and CRISPR Therapeutics are the first companies to seek FDA clearance for a gene-editing therapy.
Vertex Pharmaceuticals and CRISPR Therapeutics have gotten closer to introducing exagamglogene autotemcel (exa-cel), a one-time treatment for sickle cell disease (SCD) and transfusion-dependent beta-thalassemia (TDT). The two collaborators have submitted a marketing application for exagamglogene autotemcel (exa-cel, formerly CTX001) for the blood disorders, sickle cell disease (SCD) and beta-thalassemia, just ten years after the Nobel Prize-winning CRISPR/Cas9 gene-editing technology first emerged from the lab and seven years after the first CRISPR therapy began clinical testing.
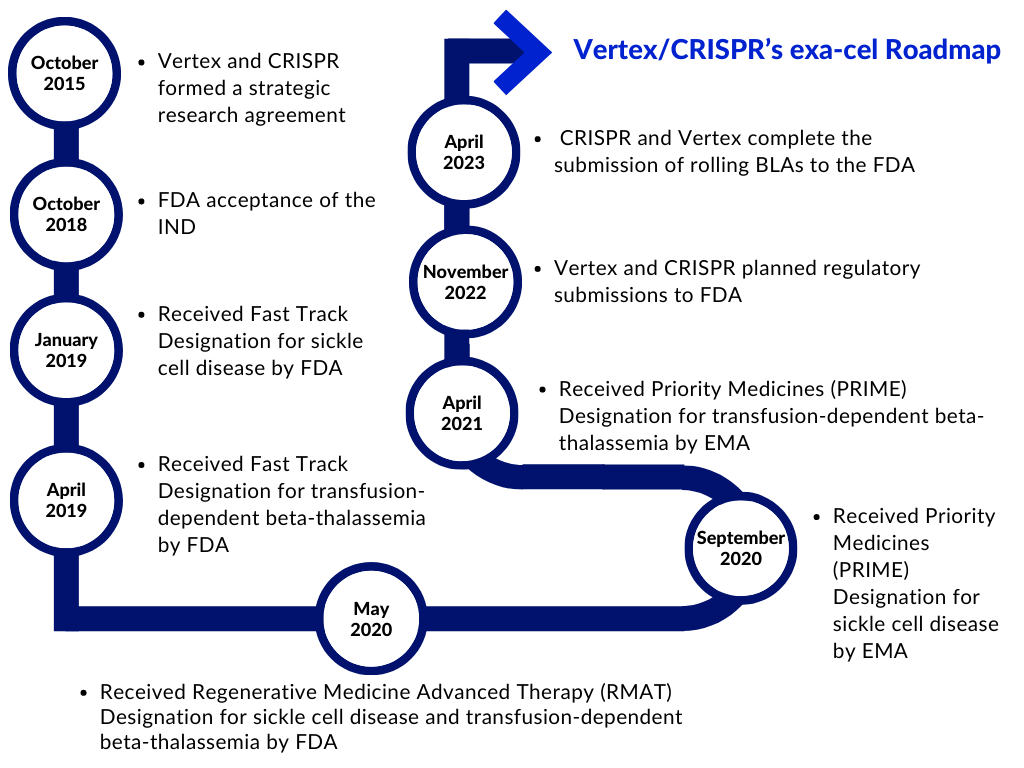
Vertex and CRISPR Therapeutics formed a strategic research agreement in 2015 to find and develop potential novel treatments targeting human disease’s underlying genetic causes using CRISPR/Cas9. Exa-cel is the first potential therapeutic to emerge from the collaborative research program. Vertex now oversees the global development, manufacture, and commercialization of exa-cel under a modified partnership agreement, and the company distributes program expenses and profits globally 60/40 with CRISPR Therapeutics.
Downloads
Article in PDF
Recent Articles
- Abingworth & Alebund’s Finacial Closing; Pfizer/BioNTech COVID-19 Vaccine Expanded Use...
- IntraBio’s AQNEURSA Niemann-Pick Disease Approval; FDA Approves Novel Schizophrenia Drug After 35...
- Reversing Ischemia with Gene therapy
- FDA Grants Orphan Status to MDL-101 for LAMA2-CMD; Pfizer’s ABRYSVO Approved for High-Risk Adults...
- Opportunities and Challenges for Cell and Gene Therapies
Vertex and CRISPR began preparing the FDA application in November last year and have requested a priority review from the FDA to decrease the review period at the agency to eight months from submission rather than 12 months. Vertex’s most advanced contender outside of its cystic fibrosis program, the therapy is on track to be the first CRISPR-based gene editing therapeutic authorized by the FDA. The BLAs submission was backed up by data from two ongoing Phase III studies, CLIMB-111 and CLIMB-121, as well as a long-term follow-up study, CLIMB-131. Data from the Phase III studies were most recently reported in December 2022 at the American Society of Hematology (ASH) Annual Meeting and Exposition. Vertex is conducting five studies on exa-cel: the Phase I/II/III open-label CLIMB-111 and CLIMB-121 trials, the Phase III open-label CLIMB-141 and CLIMB-151 trials, and the long-term open-label CLIMB-131 trial.
Patients who participate in these trials will have their own hematopoietic stem and progenitor cells extracted from their peripheral blood. CRISPR/Cas9 technology will be used to modify the patient’s cells. The altered cells, known as exa-cel, will then be injected back into the patient as part of an autologous hematopoietic stem cell transplant (HSCT), a procedure that requires myeloablative busulfan conditioning. Patients receiving HSCT may also experience side effects (varying from mild to severe) unrelated to exa-cel treatment. Patients will be observed at first to see when the altered cells start producing mature blood cells, a process known as engraftment. Patients will be observed after engraftment to track the impact of exa-cel on numerous disease parameters and for safety.
“The completion of our exa-cel global regulatory filings is a historic milestone,” said Carmen Bozic, M.D., Vertex Pharmaceuticals’ Executive Vice President, Global Medicines Development and Medical Affairs, and Chief Medical Officer. “We’d like to thank the clinical trial participants, the sickle cell and beta thalassemia communities, and the physicians, nurses, coordinators, caregivers, and friends who have supported them.”
“Within a decade, we went from discovering the CRISPR platform to the first regulatory filings for a CRISPR-based therapy, demonstrating the transformative nature of CRISPR technology,” said Phuong Khanh (P.K.) Morrow, M.D., FACP, Chief Medical Officer at CRISPR Therapeutics.
In the U.S., exa-cel has been granted Regenerative Medicine Advanced Therapy (RMAT), Fast Track, Orphan Drug, and Rare Pediatric Disease designations from the FDA for both sickle cell disease and transfusion-dependent beta-thalassemia as a symbol of the potential exa-cel has to change the space. In Europe, the Marketing Authorization Applications (MAAs), for exa-cel were submitted in December 2022 and validated by the European Medicines Agency (EMA) and the Medicines and Healthcare Products Regulatory Agency (MHRA) in January 2023. In the EU, exa-cel has been granted Orphan Drug Designation from the European Commission and Priority Medicines (PRIME) designation from the EMA for both sickle cell disease and transfusion-dependent beta-thalassemia. In the U.K., exa-cel has also been granted an Innovation Passport under the Innovative Licensing and Access Pathway (ILAP) from the MHRA.
Exa-cel, formerly known as CTX001, is an investigational, autologous, ex vivo CRISPR/Cas9 gene-edited therapy for patients with sickle cell disease or transfusion-dependent beta-thalassemia in which a patient’s own hematopoietic stem cells are edited to produce high levels of fetal hemoglobin in red blood cells. Sickle cell disease and transfusion-dependent beta-thalassemia are caused by beta-globin gene mutations, which generate a protein essential for hemoglobin function. The defective protein results in aberrant hemoglobin, which causes the anemia found in both disorders.
Vertex and CRISPR hope to address the fundamental cause of sickle cell disease and transfusion-dependent beta-thalassemia using exa-cel. The candidate is an autologous and ex vivo gene-edited therapy that edits a patient’s own hematopoietic stem cells to create high quantities of fetal hemoglobin using the CRISPR/Cas9 technology. This protein version transports oxygen more efficiently than adult hemoglobin, and increased amounts of the fetal protein can reduce anemia, potentially reducing the need for blood transfusions and alleviating painful and debilitating episodes in sickle cell disease patients.

Vertex has built the necessary manufacturing capacity in preparation for the launch, as gene therapy pricing has always been an issue. Treatment expenditures for severe sickle cell disease patients in the United States run between USD 4 million and USD 6 million over a lifetime. Even if the companies price exa-cel at the lower end of that range, the one-time medicine would surpass the USD 3.5 million drug-price record recently set by CSL’s hemophilia B gene therapy Hemgenix.
If approved, exa-cel will be the first gene editing medicine based on the Nobel Prize-winning CRISPR technology. However, it will certainly have to compete with bluebird Bio’s competitor gene therapy, lovo-cel in sickle cell disease and Zynteglo in beta-thalassemia. Zynteglo, like all one-time, gene-directed therapy, has a high price tag of USD 2.8 million.
Before Zynteglo’s clearance in the United States in August, bluebird had exited the European market after failing to achieve reimbursement deals with local governments. Vertex is currently talking to commercial and government payers and policymakers in the United States and Europe, hoping not to replicate bluebird’s experience. When exa-cel is approved, the company intends to provide widespread patient access and reimbursement agreements.
According to Stuart A. Arbuckle, Executive Vice President and Chief Operating Officer, Vertex has met with every state Medicaid agency in the United States, as well as approximately 150 commercial payers in the United States and many health economic evaluation authorities in Europe, including the United Kingdom’s National Institute for Health and Care Excellence. So far, payers consider exa-cel’s clinical results to be “highly impactful,” particularly in terms of reducing vaso-occlusive events and hospitalizations, he said.
Exa-cel is not the same as bluebird medications. Rather than employing viral vectors to transmit a functioning HBB gene, the Vertex-CRISPR therapy employs CRISPR to switch off the suppression of fetal hemoglobin and boost the amount of healthy hemoglobin in red blood cells. Vertex estimates that around 32,000 patients in the United States may be eligible for treatment with exa-cel if approved but has yet to comment on price plans or provide sales figures, citing uncertain approval timelines.
FAQs
Sickle Cell Disease is a group of inherited red blood cell disorders that affects hemoglobin, the protein that carries oxygen through the body. Sickle cells are rapidly destroyed in the bodies of patients suffering from the condition, resulting in anemia, which is why it is generally referred to as sickle cell anemia.
Sickle cell anemia symptoms commonly begin around the age of 6 months. They differ from person to person and may alter over time. Common sickle cell disease symptoms include anemia, pain episodes, hand and foot swelling, vision issues, and others.
A blood and bone marrow transplant is now some patients’ sole cure and sickle cell disease treatment option. Following an early sickle cell disease diagnosis, the healthcare professional may offer medications or transfusions to treat problems such as chronic pain.
Beta-thalassemia is a hereditary disorder that causes anemia caused by a lack of healthy red blood cells (RBCs). Without enough healthy RBCs, cells, and tissues throughout the body lack the oxygen required to operate.
Patients with severe beta-thalassemia require lifelong beta-thalassemia treatment to avoid and manage the disease’s clinical implications. Long-term treatment adherence is required to maintain blood composition levels. The Thalassemia International Federation has published comprehensive management guidelines for both TDT and NTDT patients, which are widely available.

Downloads
Article in PDF
Recent Articles
- DelveInsight’s Dermatology based Gene Therapy Reports
- Metachromatic Leukodystrophy (MLD): A Rare Indication with great unmet medical need
- What are your views on Gene Therapy?
- FDA rejects BioMarin’s Valoctocogene Roxaparvovec; J&J inks $6.5B deal; Alzheon bags $...
- Cell and Gene Therapies for Diabetes Treatment: A Permanent Cure for Patients?